Ignm: a Database of Protein Functional Motions Based on Gaussian Network Model Lee-Wei Yang1,∗, Xiong Liu2, Christopher J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METABOLIC EVOLUTION in GALDIERIA SULPHURARIA By
METABOLIC EVOLUTION IN GALDIERIA SULPHURARIA By CHAD M. TERNES Bachelor of Science in Botany Oklahoma State University Stillwater, Oklahoma 2009 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY May, 2015 METABOLIC EVOLUTION IN GALDIERIA SUPHURARIA Dissertation Approved: Dr. Gerald Schoenknecht Dissertation Adviser Dr. David Meinke Dr. Andrew Doust Dr. Patricia Canaan ii Name: CHAD M. TERNES Date of Degree: MAY, 2015 Title of Study: METABOLIC EVOLUTION IN GALDIERIA SULPHURARIA Major Field: PLANT SCIENCE Abstract: The thermoacidophilic, unicellular, red alga Galdieria sulphuraria possesses characteristics, including salt and heavy metal tolerance, unsurpassed by any other alga. Like most plastid bearing eukaryotes, G. sulphuraria can grow photoautotrophically. Additionally, it can also grow solely as a heterotroph, which results in the cessation of photosynthetic pigment biosynthesis. The ability to grow heterotrophically is likely correlated with G. sulphuraria ’s broad capacity for carbon metabolism, which rivals that of fungi. Annotation of the metabolic pathways encoded by the genome of G. sulphuraria revealed several pathways that are uncharacteristic for plants and algae, even red algae. Phylogenetic analyses of the enzymes underlying the metabolic pathways suggest multiple instances of horizontal gene transfer, in addition to endosymbiotic gene transfer and conservation through ancestry. Although some metabolic pathways as a whole appear to be retained through ancestry, genes encoding individual enzymes within a pathway were substituted by genes that were acquired horizontally from other domains of life. Thus, metabolic pathways in G. sulphuraria appear to be composed of a ‘metabolic patchwork’, underscored by a mosaic of genes resulting from multiple evolutionary processes. -

Discovery of Novel Bacterial Queuine Salvage Enzymes and Pathways in Human Pathogens
Discovery of novel bacterial queuine salvage enzymes and pathways in human pathogens a,1 b,1 c,1 b d a Yifeng Yuan , Rémi Zallot , Tyler L. Grove , Daniel J. Payan , Isabelle Martin-Verstraete , Sara Sepic´ , Seetharamsingh Balamkundue, Ramesh Neelakandane, Vinod K. Gadie, Chuan-Fa Liue, Manal A. Swairjof,g, Peter C. Dedone,h,i, Steven C. Almoc, John A. Gerltb,j,k, and Valérie de Crécy-Lagarda,l,2 aDepartment of Microbiology and Cell Science, University of Florida, Gainesville, FL 32611; bInstitute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, IL 61801; cDepartment of Biochemistry, Albert Einstein College of Medicine, Bronx, NY 10461; dLaboratoire de Pathogénèse des Bactéries Anaérobies, Institut Pasteur et Université de Paris, F-75015 Paris, France; eSingapore-MIT Alliance for Research and Technology, Infectious Disease Interdisciplinary Research Group, 138602 Singapore, Singapore; fDepartment of Chemistry and Biochemistry, San Diego State University, San Diego, CA 92182; gThe Viral Information Institute, San Diego State University, San Diego, CA 92182; hDepartment of Biological Engineering and Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139; iCenter for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139; jDepartment of Biochemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801; kDepartment of Chemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801; and lUniversity of Florida Genetics Institute, Gainesville, FL 32610 Edited by Tina M. Henkin, The Ohio State University, Columbus, OH, and approved August 1, 2019 (received for review June 16, 2019) Queuosine (Q) is a complex tRNA modification widespread in 1A. The TGT enzyme, which is responsible for the base ex- eukaryotes and bacteria that contributes to the efficiency and accuracy change, is the signature enzyme in the Q biosynthesis pathway. -

Downloaded As a Text File, Is Completely Dynamic
BMC Bioinformatics BioMed Central Database Open Access ORENZA: a web resource for studying ORphan ENZyme activities Olivier Lespinet and Bernard Labedan* Address: Institut de Génétique et Microbiologie, CNRS UMR 8621, Université Paris-Sud, Bâtiment 400, 91405 Orsay Cedex, France Email: Olivier Lespinet - [email protected]; Bernard Labedan* - [email protected] * Corresponding author Published: 06 October 2006 Received: 25 July 2006 Accepted: 06 October 2006 BMC Bioinformatics 2006, 7:436 doi:10.1186/1471-2105-7-436 This article is available from: http://www.biomedcentral.com/1471-2105/7/436 © 2006 Lespinet and Labedan; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Despite the current availability of several hundreds of thousands of amino acid sequences, more than 36% of the enzyme activities (EC numbers) defined by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) are not associated with any amino acid sequence in major public databases. This wide gap separating knowledge of biochemical function and sequence information is found for nearly all classes of enzymes. Thus, there is an urgent need to explore these sequence-less EC numbers, in order to progressively close this gap. Description: We designed ORENZA, a PostgreSQL database of ORphan ENZyme Activities, to collate information about the EC numbers defined by the NC-IUBMB with specific emphasis on orphan enzyme activities. -

(12) United States Patent (10) Patent No.: US 9,689,046 B2 Mayall Et Al
USOO9689046B2 (12) United States Patent (10) Patent No.: US 9,689,046 B2 Mayall et al. (45) Date of Patent: Jun. 27, 2017 (54) SYSTEM AND METHODS FOR THE FOREIGN PATENT DOCUMENTS DETECTION OF MULTIPLE CHEMICAL WO O125472 A1 4/2001 COMPOUNDS WO O169245 A2 9, 2001 (71) Applicants: Robert Matthew Mayall, Calgary (CA); Emily Candice Hicks, Calgary OTHER PUBLICATIONS (CA); Margaret Mary-Flora Bebeselea, A. et al., “Electrochemical Degradation and Determina Renaud-Young, Calgary (CA); David tion of 4-Nitrophenol Using Multiple Pulsed Amperometry at Christopher Lloyd, Calgary (CA); Lisa Graphite Based Electrodes', Chem. Bull. “Politehnica” Univ. Kara Oberding, Calgary (CA); Iain (Timisoara), vol. 53(67), 1-2, 2008. Fraser Scotney George, Calgary (CA) Ben-Yoav. H. et al., “A whole cell electrochemical biosensor for water genotoxicity bio-detection”. Electrochimica Acta, 2009, 54(25), 6113-6118. (72) Inventors: Robert Matthew Mayall, Calgary Biran, I. et al., “On-line monitoring of gene expression'. Microbi (CA); Emily Candice Hicks, Calgary ology (Reading, England), 1999, 145 (Pt 8), 2129-2133. (CA); Margaret Mary-Flora Da Silva, P.S. et al., “Electrochemical Behavior of Hydroquinone Renaud-Young, Calgary (CA); David and Catechol at a Silsesquioxane-Modified Carbon Paste Elec trode'. J. Braz. Chem. Soc., vol. 24, No. 4, 695-699, 2013. Christopher Lloyd, Calgary (CA); Lisa Enache, T. A. & Oliveira-Brett, A. M., "Phenol and Para-Substituted Kara Oberding, Calgary (CA); Iain Phenols Electrochemical Oxidation Pathways”, Journal of Fraser Scotney George, Calgary (CA) Electroanalytical Chemistry, 2011, 1-35. Etesami, M. et al., “Electrooxidation of hydroquinone on simply prepared Au-Pt bimetallic nanoparticles'. Science China, Chem (73) Assignee: FREDSENSE TECHNOLOGIES istry, vol. -

Functional Annotation of Uncharacterized Enzymes in Yeast
Functional Annotation of Uncharacterized Enzymes in Saccharomyces cerevisiae by Julia Ann Hanchard A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Julia Hanchard 2019 Abstract Functional Annotation of Uncharacterized Enzymes in Saccharomyces cerevisiae Julia Hanchard Doctor of Philosophy, 2019 Department of Molecular Genetics University of Toronto In the post-genomic era, clinicians and scientists are increasingly reliant on interpretation of variants in metabolic genes for determining pathogenicity. These interpretations depend on functional annotation of the roles genes provide in metabolism, an annotation that is far from complete. I embarked on a journey of enzyme discovery to fill gaps in our knowledge of metabolism in the budding yeast, Saccharomyces cerevisiae. I carried out a genetic and metabolomic screen of 120 uncharacterized candidate enzyme encoding genes that comprised my master’s thesis. This dissertation describes my work in ascribing function to two distinct enzymes, Das2 and Tda5. Throughout my study I have found that Das2 is a novel uridine/cytidine kinase that functions in concert with a second minor uridine kinase, Urk1. These two enzymes are interdependent and in turn depend on a third enzyme, the major uracil phosphoribosyl transferase, Fur1 for stability. These three enzymes form a complex that is essential to wild-type pyrimidine salvage. As I aimed to elucidate the function of Tda5, I discovered that this uncharacterized enzyme is essential to growth. Loss of function mutations in TDA5 are alleviated by de-repression of its sporulation specific paralog, Ydl114w, and when YDL114W is deleted, tda5Δ is rescued by hypomorphic mutations in the ergosterol biosynthetic pathway. -

Coupled Nucleoside Phosphorylase Reactions in Escherichia Coli John Lewis Ott Iowa State College
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1956 Coupled nucleoside phosphorylase reactions in Escherichia coli John Lewis Ott Iowa State College Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Biochemistry Commons, and the Microbiology Commons Recommended Citation Ott, John Lewis, "Coupled nucleoside phosphorylase reactions in Escherichia coli " (1956). Retrospective Theses and Dissertations. 13758. https://lib.dr.iastate.edu/rtd/13758 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. NOTE TO USERS This reproduction is the best copy available. UMI COUPLED NUCLEOSIDE PHOSPHORYLASE REACTIONS IN ESCHERICHIA COLI / by John Lewis Ott A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Physlolgglcal Bacteriology Approved: Signature was redacted for privacy. In Charge of Major Work Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Dean of Graduate College Iowa State College 1956 UMI Number: DP12892 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -
Generate Metabolic Map Poster
Authors: Zheng Zhao, Delft University of Technology Marcel A. van den Broek, Delft University of Technology S. Aljoscha Wahl, Delft University of Technology Wilbert H. Heijne, DSM Biotechnology Center Roel A. Bovenberg, DSM Biotechnology Center Joseph J. Heijnen, Delft University of Technology An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Marco A. van den Berg, DSM Biotechnology Center Peter J.T. Verheijen, Delft University of Technology Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. PchrCyc: Penicillium rubens Wisconsin 54-1255 Cellular Overview Connections between pathways are omitted for legibility. Liang Wu, DSM Biotechnology Center Walter M. van Gulik, Delft University of Technology L-quinate phosphate a sugar a sugar a sugar a sugar multidrug multidrug a dicarboxylate phosphate a proteinogenic 2+ 2+ + met met nicotinate Mg Mg a cation a cation K + L-fucose L-fucose L-quinate L-quinate L-quinate ammonium UDP ammonium ammonium H O pro met amino acid a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar K oxaloacetate L-carnitine L-carnitine L-carnitine 2 phosphate quinic acid brain-specific hypothetical hypothetical hypothetical hypothetical -
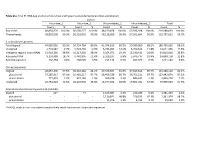
Table S1a. Trial #1 RNA-Seq Analysis of Virus-Free and Hypovirus
Table S1a. Trial #1 RNA-Seq analysis of virus-free and hypovirus-transfected Sclerotinia sclerotiorum Library Virus-free_1 Virus-free_2 Virus-infected_1 Virus-infected_2 Total Count % Count % Count % Count % Count % Raw reads 48,854,170 100.0% 56,568,277 100.0% 48,169,458 100.0% 57,396,928 100.0% 210,988,833 100.0% Filtered reads 48,810,008 99.9% 56,518,010 99.9% 48,118,009 99.9% 57,341,604 99.9% 210,787,631 99.9% S. sclerotiorum genome Total aligned 44,085,026 90.3% 50,724,764 89.8% 41,374,916 86.0% 50,580,960 88.2% 186,765,666 88.6% Unaligned 4,724,982 9.7% 5,793,246 10.3% 6,743,093 14.0% 6,760,644 11.8% 24,021,965 11.5% Intergenic regions (non-rRNA) 13,918,285 28.5% 16,317,039 28.9% 9,354,071 19.4% 15,230,421 26.6% 54,819,816 25.8% Ribosomal RNA 9,150,409 18.7% 6,442,905 11.4% 3,329,107 6.8% 5,976,737 10.4% 24,899,158 11.8% Retrotransposons 202,264 0.4% 268,505 0.5% 251,134 0.5% 499,377 0.9% 1,221,280 0.6% Coding sequences Aligned 28,057,390 57.5% 36,310,362 64.2% 29,370,507 61.0% 37,649,843 65.7% 131,388,102 62.1% plus strand 27,285,917 97.3% 35,481,217 97.7% 28,409,529 96.7% 36,766,211 97.7% 127,942,874 97.3% minus strand 771,473 2.7% 829,145 2.3% 960,978 3.3% 883,632 2.3% 3,445,228 2.7% Unaligned 20,752,618 42.5% 20,207,648 35.8% 18,747,502 39.0% 19,691,761 34.3% 79,399,529 37.9% Sclerotinia sclerotiorum hypovirus 2L (SsHV2L) Aligned 62* - 77 - 1,165,087 2.4% 216,286 0.4% 1,381,450 1.4% plus strand - - - - 1,150,841 98.8% 210,533 97.3% 1,361,374 98.1% minus strand - - - - 14,246 1.2% 5,753 2.7% 19,999 1.9% *SsHV2L reads in non-inoculated samples likely result from errors in barcode sequences. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Study of Nucleoside Degrading Enzyme Activities in Bean, Organic Bean, Okra, Organic Okra, Squash and Organic Squash
STUDY OF NUCLEOSIDE DEGRADING ENZYME ACTIVITIES IN BEAN, ORGANIC BEAN, OKRA, ORGANIC OKRA, SQUASH AND ORGANIC SQUASH by Shafiqa A. Alshaiban A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Chemistry Middle Tennessee State University August 2016 Thesis Committee: Dr. Paul C. Kline, Chair Dr. Andrew Burden Dr. Anthony Farone I dedicate this research to my parents, my sisters, and my brothers. I love you all. ii ACKNOWLEDGEMENTS I would like to sincerely thank Dr. Paul Kline for the support and guidance he has offered throughout the entirety of the research and thesis writing process. I also wish to thank my committee members, Dr. Donald A. Burden and Dr. Anthony Farone for their advice and willingness to read my work. I would like to thank all the staff and faculty members for their valuable support. Finally, I would like to express my thanks to my family and my friends for their support and love. Special thanks to my loving parents, my brothers and my sisters for their prayer, concern and kind words over the years. iii ABSTRACT Pyrimidine and purine nucleotide metabolism are essential for development and growth of all organisms. Nucleoside degradation reactions have been found in virtually all organisms. Many enzymes are involved in the degradation and salvage of nucleotides, nucleobases and nucleosides. Deaminases contribute in interconversion of one nucleoside into another by removing amino groups from the base. Nucleoside hydrolase is a glycosidase that catalyzes the cleavage of the N-glycosidic bond in nucleosides to facilitate recycling of nucleobases. -
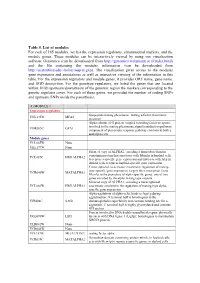
Table 5. List of Modules for Each of 165 Modules, We List the Expression Regulators, Chromosomal Markers, and the Module Genes
Table 5. List of modules For each of 165 modules, we list the expression regulators, chromosomal markers, and the module genes. These modules can be interactively viewed by using our visualization software Genomica (can be downloaded from http://genomica.weizmann.ac.il/index.html) and the file containing the modules information (can be downloaded from http://ai.stanford.edu/~koller/seqvar.gxp). The visualization gives access to the modules gene expression and annotations as well as interactive viewing of the information in this table. For the expression regulators and module genes, it provides ORF name, gene name, and SGD desciprtion. For the genotype regulators, we listed the genes that are located within 10 kb upstream/downstream of the genomic region the markers corresponding to the genetic regulator cover. For each of these genes, we provided the number of coding SNPs and upstream SNPs inside the parenthesis. (1) MODULE 1 Expression regulators lipopeptide mating pheromone; mating a-factor pheromone YNL145W MFA2 precursor Alpha subunit of G protein coupled to mating factor receptors, involved in the mating pheromone signal transduction pathway; YHR005C GPA1 component of pheromone response pathway common to both a and alpha cells Module genes YCL065W None YKL177W None Silenced copy of ALPHA2, encoding a homeobox-domain containing protein that associates with Mcm1p in haploid cells YCL067C HMLALPHA2 to repress a-specific gene expression and interacts with A1p in diploid cells to repress haploid-specific gene expression Transcriptional -

Bombyx Mori) Strains of Variable Silk Yield
Article Identification of Genes that Control Silk Yield by RNA Sequencing Analysis of Silkworm (Bombyx mori) Strains of Variable Silk Yield Yue Luan, Weidong Zuo, Chunlin Li, Rui Gao, Hao Zhang, Xiaoling Tong, Minjin Han, Hai Hu, Cheng Lu and Fangyin Dai * State Key Laboratory of Silkworm Genome Biology, Key Laboratory of Sericultural Biology and Genetic Breeding, Ministry of Agriculture, College of Biotechnology, Southwest University, Chongqing 400715, China; [email protected] (Y.L.); [email protected] (W.Z.); [email protected] (C.L.); [email protected] (R.G.); [email protected] (H.Z.); [email protected] (X.T.); [email protected] (M.H.); [email protected] (H.C.); [email protected] (C.L.) * Correspondence: [email protected]; Tel.: +86-23-68250793 Received: 27 October 2018; Accepted: 15 November 2018; Published: 22 November 2018 Abstract: Silk is an important natural fiber of high economic value, and thus genetic study of the silkworm is a major area of research. Transcriptome analysis can provide guidance for genetic studies of silk yield traits. In this study, we performed a transcriptome comparison using multiple silkworms with different silk yields. A total of 22 common differentially expressed genes (DEGs) were identified in multiple strains and were mainly involved in metabolic pathways. Among these, seven significant common DEGs were verified by quantitative reverse transcription polymerase chain reaction, and the results coincided with the findings generated by RNA sequencing. Association analysis showed that BGIBMGA003330 and BGIBMGA005780 are significantly associated with cocoon shell weight and encode uridine nucleosidase and small heat shock protein, respectively. Functional annotation of these genes suggest that these play a role in silkworm silk gland development or silk protein synthesis.