Cdk2 Catalytic Activity Is Essential for Meiotic Cell Division in Vivo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Reciprocal Effort Towards Better Approaches for Drug Discovery
Acta Pharmacologica Sinica (2013) 34: 765–776 npg © 2013 CPS and SIMM All rights reserved 1671-4083/13 $32.00 www.nature.com/aps Review iPSCs and small molecules: a reciprocal effort towards better approaches for drug discovery Ru ZHANG1, Li-hong ZHANG2, Xin XIE1, 2, * 1Laboratory of Receptor-based Bio-medicine, Shanghai Key Laboratory of Signaling and Disease Research, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China; 2CAS Key Laboratory of Receptor Research, the National Center for Drug Screening, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China The revolutionary induced pluripotent stem cell (iPSC) technology provides a new path for cell replacement therapies and drug screen- ing. Patient-specific iPSCs and subsequent differentiated cells manifesting disease phenotypes will finally position human disease pathology at the core of drug discovery. Cells used to test the toxic effects of drugs can also be generated from normal iPSCs and pro- vide a much more accurate and cost-effective system than many animal models. Here, we highlight the recent progress in iPSC-based cell therapy, disease modeling and drug evaluations. In addition, we discuss the use of small molecule drugs to improve the genera- tion of iPSCs and understand the reprogramming mechanism. It is foreseeable that the interplay between iPSC technology and small molecule compounds will push forward the applications of iPSC-based therapy and screening systems in the real world and eventually revolutionize the methods used to treat diseases. Keywords: induced pluripotent stem cells (iPSCs); disease modeling; drug screening; toxicity evaluation; cell replacement therapy; small molecules; drug development Acta Pharmacologica Sinica (2013) 34: 765–776; doi: 10.1038/aps.2013.21; published online 22 Apr 2013 Introduction his/her own iPSCs[1–3]. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

UC San Francisco Electronic Theses and Dissertations
UCSF UC San Francisco Electronic Theses and Dissertations Title Chemical genetic approaches to study protein kinase signaling pathways Permalink https://escholarship.org/uc/item/0sn1j4nk Author Hertz, Nicholas T. Publication Date 2013 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Copyright 2013 by Nicholas T. Hertz ii Acknowledgements I would like to thank Kevan for his support, insight, encouragement and enthusiasm for science. I would also like to thank Al for all of his support and encouragement. My parents were instrumental in me getting to this point and I want to thank them for their continued support and my wife Janel for everything. Part of this thesis is a reproduction of material previously published, and contains contributions from collaborators listed therein. Chapter 1 is reproduced in part with permission from: Hertz, N.T., Wang, B.T., Allen, J.J., Zhang, C., Dar, A.C., Burlingame, A.L., and Shokat, K.M., Chemical genetic approach for kinase-substrate mapping by covalent capture of thiophosphopeptides and analysis by mass spectrometry, Current Protocols in Chemical Biology. 2010, February; 2:(15-36). Chapter 2 is reproduced in part with permission from: Ultanir, S.K.*, Hertz, N.T.*, Li, G., Ge1, W.P., Burlingame, A.L., Pleasure, S.J., Shokat, K.M., Jan, L.Y., and Jan, Y., Chemical genetic identification of NDR1/2 kinase substrates AAK1 and Rabin8 uncovers their roles in controlling dendrite arborization and synapse maturation. Neuron. 2012, March 22; 73:1127-42. Chapter 3 is reproduced in part with permission from: Hengeveld, R.C.C.*, Hertz, N.T.*, Vromans, M.J.M., Zhang, C., Burlingame, A.L., Shokat, K.M., Lens, S.M.A., Development of a chemical genetic approach for human Aurora B kinase identifies novel substrates of the chromosomal passenger complex. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Rectifier of Aberrant Mrna Splicing Recovers Trna Modification in Familial Dysautonomia
Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia Mayumi Yoshidaa, Naoyuki Kataokab,1, Kenjyo Miyauchic, Kenji Ohea,d, Kei Iidaa,e, Suguru Yoshidaf, Takayuki Nojimag, Yukiko Okunoa,e, Hiroshi Onogia,h, Tomomi Usuii, Akihide Takeuchia, Takamitsu Hosoyaf, Tsutomu Suzukic, and Masatoshi Hagiwaraa,1 aDepartment of Anatomy and Developmental Biology, Kyoto University Graduate School of Medicine, Kyoto 606-8501, Japan; bLaboratory for Malignancy Control Research, Medical Innovation Center, Kyoto University Graduate School of Medicine, Kyoto 606-8507, Japan; cDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Tokyo 113-8510, Japan; dKyoto University Graduate School of Medicine, Kyoto 606-8501, Japan; eMedical Research Support Center, Kyoto University Graduate School of Medicine, Kyoto 606-8507, Japan; fLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University, Tokyo 101-0062, Japan; gSir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, United Kingdom; hKinoPharma, Inc., Tokyo 154-0024, Japan; and iLaboratory of Gene Expression, School of Biomedical Science, Tokyo Medical and Dental University, Tokyo 113-8510, Japan Edited by Gideon Dreyfuss, University of Pennsylvania, Philadelphia, PA, and approved December 30, 2014 (received for review August 12, 2014) Familial dysautonomia (FD), a hereditary sensory and autonomic pneumonia, vomiting/dysautonomic crisis, gastrointestinal dys- neuropathy, is caused by missplicing of exon 20, resulting from an function, decreased sensitivity to pain and temperature, and intronic mutation in the inhibitor of kappa light polypeptide gene defective lacrimation. FD is a very common disorder in the enhancer in B cells, kinase complex-associated protein (IKBKAP) Ashkenazi Jewish population, with a carrier frequency of 1 in 27. -

Whole Transcriptomic Expression Differences in EBV Immortalized Versus Primary B-Cells
W&M ScholarWorks Undergraduate Honors Theses Theses, Dissertations, & Master Projects 12-2010 Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells Dolores Huberts College of William and Mary Follow this and additional works at: https://scholarworks.wm.edu/honorstheses Part of the Biology Commons Recommended Citation Huberts, Dolores, "Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B- Cells" (2010). Undergraduate Honors Theses. Paper 347. https://scholarworks.wm.edu/honorstheses/347 This Honors Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Undergraduate Honors Theses by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells A thesis submitted in partial fulfillment of the requirement for the degree of Bachelor of Science with Honors in Biology from the College of William and Mary in Virginia By Dolores Huberts Accepted for Honors ________________________________________ Lizabeth A. Allison, Director ________________________________________ Matthew Wawersik ________________________________________ Drew LaMar ________________________________________ Beverly Sher Williamsburg, Virginia December 17, 2010 ABSTRACT The Epstein–Barr Virus (EBV) is a human gamma herpes virus that infects more than 90% of the human population worldwide. It is commonly known in the US as the cause of Infectious Mononucleosis, and around the world as the cause of nasopharyngeal carcinoma and malignant lymphomas such as non-Hodgkin lymphoma, endemic Burkett’s lymphoma and Hodgkin lymphoma. Additionally, the EBV is used to immortalize cells to create cell lines for in-vitro studies. -

Inherited Neuropathies: Emerging Genetic Therapies
5th Congress of the European Academy of Neurology Oslo, Norway, June 29 - July 2, 2019 Teaching Course 3 EAN/PNS: Novel approach in the treatment of neuropathy (Level3) Inherited neuropathies: Emerging genetic therapies Mary M. Reilly London, United Kingdom Email: [email protected] 09/07/2019 Mary M Reilly MRC centre for Neuromuscular Diseases, Institute of Neurology, Queen Square, London, UK. IONIS TTR trial Consultancy Alnylam Inflectis Acceleron Akcea Myotherix 1 09/07/2019 1. Introduction 2. Barriers to therapy development 3. Classification of therapies 4. Emerging therapies 2. Barriers to therapy development 3. Classification of therapies 4. Emerging therapies 2 09/07/2019 Charcot Marie Tooth disease 1. Sole / primary e.g. CMT 2. Part of multisystem disorder 3 09/07/2019 2. Part of multisystem disorder 1. Charcot-Marie-Tooth disease (CMT) 2. Hereditary Neuropathy with liability to pressure palsies (HNPP) 3. Hereditary sensory neuropathies (HSN / HSAN) 4. Distal hereditary motor neuropathies (HMN) 4 09/07/2019 5 09/07/2019 17p, LITAF, DYNC1H1, BICD2, REEP1, HSPB3, EGR2, FBLN5, SLC5A7, FBXO38, SETX, PMP22, PMP2 DCTN1, 7q34, WARS, MFN2, NEFL, MYH14 SPTLC1, GDAP1, MPZ, GJB1, SPTLC2, LRSAM1, YARS, INF2, ATL1, NEFH, DRP2, Xq27.1, MARS, ATL3, DNM2, KIF5A, DNMT1, GNB4 ATP1A1, SCN11A, VCP, TFG, SCN9A DHTKD1, TUBB3, NAGLU, DCAF8, PRNP, DGAT2, PDK3 COL6A5, RNF170 MORC2, HSPB8, HSPB1, TRPV4, GARS, BSCL2 AARS, HARS, CHCHD10 RAB7 SH3TC2, EGR2, MTMR2, NDRG1, SIGMAR1, SBF2, SBF1, CTDP1, SURF1, VRK1, ATP7A, UBA1, FGD4, FIG4, HK1, PRX, GLE1, LAS1L WNK1, LMNA, CNTNAP1, NEFL, FAM134B, PNKP, GDAP1, ADCY6 KIF1A, TRIM2, KARS, DST, SPG11, COX6A1 NTRK1, MME, PIEZO2, MCM3AP, SCN9A, SLC25A46, IKBKAP, SCO2, MPV17, PLEKHG5 PRDM12, LRSAM1, CLTCL1, C12orf65, CCT5, AIFM1, FLVCR1, PRPS1 NGF HINT1, DNAJB2, IGHMBP2 6 09/07/2019 1. -

Familial Dysautonomia Model Reveals Ikbkap Deletion Causes Apoptosis Of
Familial dysautonomia model reveals Ikbkap deletion + causes apoptosis of Pax3 progenitors and peripheral neurons Lynn Georgea,b,1, Marta Chaverraa,1, Lindsey Wolfea, Julian Thornea, Mattheson Close-Davisa, Amy Eibsa, Vickie Riojasa, Andrea Grindelandc, Miranda Orrc, George A. Carlsonc, and Frances Lefcorta,2 aDepartment of Cell Biology and Neuroscience, Montana State University, Bozeman, MT 59717; bDepartment of Biological and Physical Sciences, Montana State University Billings, Billings, MT 59101; and cMcLaughlin Research Institute, Great Falls, MT 59405 Edited by Qiufu Ma, Dana-Farber Cancer Institute, Boston, MA, and accepted by the Editorial Board October 7, 2013 (received for review May 8, 2013) Familial dysautonomia (FD) is a devastating developmental and thermoreceptors, mechanoreceptors and proprioceptors. With progressive peripheral neuropathy caused by a mutation in the gene the completion of neural crest migration, multiple steps ensue inhibitor of kappa B kinase complex-associated protein (IKBKAP). that are essential for normal PNS development, including pro- To identify the cellular and molecular mechanisms that cause FD, liferation of discrete sets of neuronal progenitor cells that derive we generated mice in which Ikbkap expression is ablated in the from different waves of migrating neural crest cells, neuronal peripheral nervous system and identify the steps in peripheral differentiation, axonogenesis, target innervation, and circuit nervous system development that are Ikbkap-dependent. We formation. FD could theoretically result from failure in any or show that Ikbkap is not required for trunk neural crest migration several of these key developmental processes. or pathfinding, nor for the formation of dorsal root or sympathetic Insight into the mechanisms that cause FD have been com- ganglia, or the adrenal medulla. -

IKAP Antibody (Pab)
21.10.2014IKAP antibody (pAb) Rabbit Anti -Human/Mouse IKK Complex Associated Protein (IKBKAP) Instruction Manual Catalog Number PK-AB718-2337 Synonyms IKAP Antibody: IKK complex-associated protein, IKBKAP Description IKAP was initially identified as a scaffold protein of the I κB kinase complex that could bind to IKK α, IKK β, NF-κB, and the NF-κB-inducing kinase (NIK), although later evidence has cast doubt on this. More recent reports show that mutations in IKAP such as a frameshift leading to a truncated protein or a missense mutation that leads to defective phosphorylation are responsible for the autosomal recessive genetic disease familial dysautonomia (FD). Reports indicating that it forms part of the RNA polymerase II transcription elongation complex suggest that this disease may be due to compromised transcription elongation. More recently, it was shown that IKAP associates with c-Jun N-terminal kinase (JNK) and could specifically enhance JNK activation induced by the upstream JNK activators MEKK1 and ASK1, indicating another possible cause for FD. At least two isoforms of IKAP are known two exist. Quantity 100 µg Source / Host Rabbit Immunogen IKAP antibody was raised against a 16 amino acid peptide from near the carboxy terminus of human IKAP. Purification Method Affinity chromatography purified via peptide column. Clone / IgG Subtype Polyclonal antibody Species Reactivity Human, Mouse Specificity Formulation Antibody is supplied in PBS containing 0.02% sodium azide. Reconstitution During shipment, small volumes of antibody will occasionally become entrapped in the seal of the product vial. For products with volumes of 200 μl or less, we recommend gently tapping the vial on a hard surface or briefly centrifuging the vial in a tabletop centrifuge to dislodge any liquid in the container’s cap. -
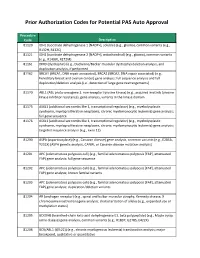
Prior Authorization Codes for Potential PAS Auto Approval
Prior Authorization Codes for Potential PAS Auto Approval Procedure Code Description 81120 IDH1 (isocitrate dehydrogenase 1 [NADP+], soluble) (e.g., glioma), common variants (e.g., R132H, R132C) 81121 IDH2 (isocitrate dehydrogenase 2 [NADP+], mitochondrial) (e.g., glioma), common variants (e.g., R140W, R172M) 81161 DMD (dystrophin) (e.g., Duchenne/Becker muscular dystrophy) deletion analysis, and duplication analysis, if performed 81162 BRCA1 (BRCA1, DNA repair associated), BRCA2 (BRCA2, DNA repair associated) (e.g., hereditary breast and ovarian cancer) gene analysis; full sequence analysis and full duplication/deletion analysis (i.e., detection of large gene rearrangements) 81170 ABL1 (ABL proto-oncogene 1, non-receptor tyrosine kinase) (e.g., acquired imatinib tyrosine kinase inhibitor resistance), gene analysis, variants in the kinase domain 81175 ASXL1 (additional sex combs like 1, transcriptional regulator) (e.g., myelodysplastic syndrome, myeloproliferative neoplasms, chronic myelomonocytic leukemia) gene analysis; full gene sequence 81176 ASXL1 (additional sex combs like 1, transcriptional regulator) (e.g., myelodysplastic syndrome, myeloproliferative neoplasms, chronic myelomonocytic leukemia) gene analysis; targeted sequence analysis (e.g., exon 12) 81200 ASPA (aspartoacylase) (e.g., Canavan disease) gene analysis, common variants (e.g., E285A, Y231X) (ASPA genetic analysis, CANW, or Canavan disease mutation analysis) 81201 APC (adenomatous polyposis coli) (e.g., familial adenomatous polyposis [FAP], attenuated FAP) gene analysis; -
Genetic Testing
genetic testing PATIENTS.ANSWERS.RESULTS.® aruplab.com/geneticstesting SEPTEMBER 2021 Information in this brochure is current as of September 2021. All content is subject to change. Please contact ARUP Client Services at 800-522-2787 with any questions or concerns. ARUP LABORATORIES As a nonprofit, academic institution of the University of Utah and its Department We believe in of Pathology, ARUP believes in collaborating, sharing knowledge, and contributing to laboratory science in ways that benefit our clients and their patients. collaborating, Our test menu is one of the broadest in the industry, encompassing more sharing than 3,000 tests, including highly specialized and esoteric assays. We offer comprehensive testing in the areas of genetics, molecular oncology, pediatrics, knowledge, and and pain management, among others. contributing ARUP’s clients include many of the nation’s university teaching hospitals to laboratory and children’s hospitals, as well as multihospital groups, major commercial laboratories, and group purchasing organizations. We believe that healthcare science in ways should be delivered as close to the patient as possible, which is why we support your efforts to be the principal healthcare provider in the communities you serve that provide by offering highly complex assays and accompanying consultative support. the best value Offering analytics, consulting, and decision support services, ARUP provides clients with the utilization management tools necessary to prosper in this time of for the patient. value-based care. Our UM+ program helps clients control utilization, reduce costs, Together, and improve patient care. In addition, ARUP is a worldwide leader in innovative laboratory research and development, led by the efforts of the ARUP Institute for ARUP and ® Clinical and Experimental Pathology .