Synthetic Analogues of Marine Natural Flavonoids As Antifouling Agents: Synthesis and Biological Evaluation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Bioactive Extraction and Nano-Encapsulation
Entry Novel Bioactive Extraction and Nano-Encapsulation Shaba Noore 1,2 , Navin Kumar Rastogi 3, Colm O’Donnell 2 and Brijesh Tiwari 1,2,* 1 Department of Food Chemistry & Technology, Teagasc Food Research Centre, Ashtown, D15 DY05 Dublin, Ireland; [email protected] 2 School of Biosystems and Food Engineering, University College Dublin, Belfield, D04 V1W8 Dublin, Ireland; [email protected] 3 Department of Food Engineering, CSIR-Central Food Technological Research Institute, Mysuru 570020, India; [email protected] * Correspondence: [email protected] Definition: An extraction technology works on the principle of two consecutive steps that involves mixture of solute with solvent and the movement of soluble compounds from the cell into the solvent and its consequent diffusion and extraction. The conventional extraction techniques are mostly based on the use of mild/high temperatures (50–90 ◦C) that can cause thermal degradation, are dependent on the mass transfer rate, being reflected on long extraction times, high costs, low extraction efficiency, with consequent low extraction yields. Due to these disadvantages, it is of interest to develop non-thermal extraction methods, such as microwave, ultrasounds, supercritical fluids (mostly using carbon dioxide, SC-CO2), and high hydrostatic pressure-assisted extractions which works on the phenomena of minimum heat exposure with reduced processing time, thereby minimizing the loss of bioactive compounds during extraction. Further, to improve the stability of these extracted compounds, nano-encapsulation is required. Nano-encapsulation is a process which forms a thin layer of protection against environmental degradation and retains the nutritional and Citation: Noore, S.; Rastogi, N.K.; functional qualities of bioactive compounds in nano-scale level capsules by employing fats, starches, O’Donnell, C.; Tiwari, B. -

Sideritis Clandestina (Bory & Chaub.) Hayek; Sideritis Raeseri Boiss
2 February 2016 EMA/HMPC/39455/2015 Committee on Herbal Medicinal Products (HMPC) Assessment report on Sideritis scardica Griseb.; Sideritis clandestina (Bory & Chaub.) Hayek; Sideritis raeseri Boiss. & Heldr.; Sideritis syriaca L., herba Final Based on Article 16d(1), Article 16f and Article 16h of Directive 2001/83/EC as amended (traditional use) Herbal substances (binomial scientific name of Sideritis scardica Griseb.; Sideritis clandestina the plant, including plant part) (Bory & Chaub.) Hayek; Sideritis raeseri Boiss. & Heldr.; Sideritis syriaca L., herba Herbal preparation Comminuted herbal substance Pharmaceutical form Comminuted herbal substance as herbal tea for oral use Rapporteur I. Chinou Peer-reviewer B. Kroes 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact An agency of the European Union © European Medicines Agency, 2016. Reproduction is authorised provided the source is acknowledged. Table of contents Table of contents ................................................................................................................... 2 1. Introduction ....................................................................................................................... 4 1.1. Description of the herbal substance(s), herbal preparation(s) or combinations thereof .. 4 1.2. Search and assessment methodology ..................................................................... 8 2. Data on -

Shilin Yang Doctor of Philosophy
PHYTOCHEMICAL STUDIES OF ARTEMISIA ANNUA L. THESIS Presented by SHILIN YANG For the Degree of DOCTOR OF PHILOSOPHY of the UNIVERSITY OF LONDON DEPARTMENT OF PHARMACOGNOSY THE SCHOOL OF PHARMACY THE UNIVERSITY OF LONDON BRUNSWICK SQUARE, LONDON WC1N 1AX ProQuest Number: U063742 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest U063742 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 ACKNOWLEDGEMENT I wish to express my sincere gratitude to Professor J.D. Phillipson and Dr. M.J.O’Neill for their supervision throughout the course of studies. I would especially like to thank Dr. M.F.Roberts for her great help. I like to thank Dr. K.C.S.C.Liu and B.C.Homeyer for their great help. My sincere thanks to Mrs.J.B.Hallsworth for her help. I am very grateful to the staff of the MS Spectroscopy Unit and NMR Unit of the School of Pharmacy, and the staff of the NMR Unit, King’s College, University of London, for running the MS and NMR spectra. -

Phenolic Profiling of Veronica Spp. Grown in Mountain, Urban and Sand Soil Environments
CORE Metadata, citation and similar papers at core.ac.uk Provided by Biblioteca Digital do IPB Phenolic profiling of Veronica spp. grown in mountain, urban and sand soil environments João C.M. Barreiraa,b,c, Maria Inês Diasa,c, Jelena Živkovićd, Dejan Stojkoviće, Marina Sokoviće, Celestino Santos-Buelgab,*, Isabel C.F.R. Ferreiraa,* aCIMO/Escola Superior Agrária, Instituto Politécnico de Bragança, Apartado 1172, 5301-855 Bragança, Portugal. bGIP-USAL, Facultad de Farmacia, Universidad de Salamanca, Campus Miguel de Unamuno, 37007 Salamanca, Spain. cREQUIMTE/Departamento de Ciências Químicas, Faculdade de Farmácia, Universidade do Porto, Rua Jorge Viterbo Ferreira, nº 228, 4050-313 Porto, Portugal. dInstitute for Medicinal Plant Research “Dr. Josif Pančić”, Tadeuša Košćuška 1, 11000 Belgrade, Serbia. eDepartment of Plant Physiology, Institute for Biological Research “Siniša Stanković”, University of Belgrade, Bulevar Despota Stefana 142, 11000 Belgrade, Serbia. * Authors to whom correspondence should be addressed (Isabel C.F.R. Ferreira; e-mail: [email protected], telephone +351273303219, fax +351273325405; e-mail: Celestino Santos- Buelga: [email protected]; telephone +34923294537; fax +34923294515). 1 Abstract Veronica (Plantaginaceae) genus is widely distributed in different habitats. Phytochemistry studies are increasing because most metabolites with pharmacological interest are obtained from plants. The phenolic compounds of V. montana, V. polita and V. spuria were tentatively identified by HPLC-DAD-ESI/MS. The phenolic profiles showed that flavones were the major compounds (V. montana: 7 phenolic acids, 5 flavones, 4 phenylethanoids and 1 isoflavone; V. polita: 10 flavones, 5 phenolic acids, 2 phenylethanoids, 1 flavonol and 1 isoflavone; V. spuria: 10 phenolic acids, 5 flavones, 2 flavonols, 2 phenylethanoids and 1 isoflavone), despite the overall predominance of flavones. -

Herbal Principles in Cosmetics Properties and Mechanisms of Action Traditional Herbal Medicines for Modern Times
Traditional Herbal Medicines for Modern Times Herbal Principles in Cosmetics Properties and Mechanisms of Action Traditional Herbal Medicines for Modern Times Each volume in this series provides academia, health sciences, and the herbal medicines industry with in-depth coverage of the herbal remedies for infectious diseases, certain medical conditions, or the plant medicines of a particular country. Series Editor: Dr. Roland Hardman Volume 1 Shengmai San, edited by Kam-Ming Ko Volume 2 Rasayana: Ayurvedic Herbs for Rejuvenation and Longevity, by H.S. Puri Volume 3 Sho-Saiko-To: (Xiao-Chai-Hu-Tang) Scientific Evaluation and Clinical Applications, by Yukio Ogihara and Masaki Aburada Volume 4 Traditional Medicinal Plants and Malaria, edited by Merlin Willcox, Gerard Bodeker, and Philippe Rasoanaivo Volume 5 Juzen-taiho-to (Shi-Quan-Da-Bu-Tang): Scientific Evaluation and Clinical Applications, edited by Haruki Yamada and Ikuo Saiki Volume 6 Traditional Medicines for Modern Times: Antidiabetic Plants, edited by Amala Soumyanath Volume 7 Bupleurum Species: Scientific Evaluation and Clinical Applications, edited by Sheng-Li Pan Traditional Herbal Medicines for Modern Times Herbal Principles in Cosmetics Properties and Mechanisms of Action Bruno Burlando, Luisella Verotta, Laura Cornara, and Elisa Bottini-Massa Cover art design by Carlo Del Vecchio. CRC Press Taylor & Francis Group 6000 Broken Sound Parkway NW, Suite 300 Boca Raton, FL 33487-2742 © 2010 by Taylor and Francis Group, LLC CRC Press is an imprint of Taylor & Francis Group, an Informa business No claim to original U.S. Government works Printed in the United States of America on acid-free paper 10 9 8 7 6 5 4 3 2 1 International Standard Book Number-13: 978-1-4398-1214-3 (Ebook-PDF) This book contains information obtained from authentic and highly regarded sources. -

Single Laboratory Validation of a Quantitative Core Shell-Based LC
Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.) Samantha Drouet, Bilal Haider Abbasi, Annie Falguieres, Waqar Ahmad, S. Sumaira, Clothilde Ferroud, Joël Doussot, Jean Vanier, Christophe Hano To cite this version: Samantha Drouet, Bilal Haider Abbasi, Annie Falguieres, Waqar Ahmad, S. Sumaira, et al.. Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Sily- marin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.). Molecules, MDPI, 2018, 23 (4), pp.904. 10.3390/molecules23040904. hal-02538464 HAL Id: hal-02538464 https://hal.archives-ouvertes.fr/hal-02538464 Submitted on 9 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License molecules Article Single Laboratory Validation of a Quantitative Core Shell-Based -

Distribution of Flavonoids Among Malvaceae Family Members – a Review
Distribution of flavonoids among Malvaceae family members – A review Vellingiri Vadivel, Sridharan Sriram, Pemaiah Brindha Centre for Advanced Research in Indian System of Medicine (CARISM), SASTRA University, Thanjavur, Tamil Nadu, India Abstract Since ancient times, Malvaceae family plant members are distributed worldwide and have been used as a folk remedy for the treatment of skin diseases, as an antifertility agent, antiseptic, and carminative. Some compounds isolated from Malvaceae members such as flavonoids, phenolic acids, and polysaccharides are considered responsible for these activities. Although the flavonoid profiles of several Malvaceae family members are REVIEW REVIEW ARTICLE investigated, the information is scattered. To understand the chemical variability and chemotaxonomic relationship among Malvaceae family members summation of their phytochemical nature is essential. Hence, this review aims to summarize the distribution of flavonoids in species of genera namely Abelmoschus, Abroma, Abutilon, Bombax, Duboscia, Gossypium, Hibiscus, Helicteres, Herissantia, Kitaibelia, Lavatera, Malva, Pavonia, Sida, Theobroma, and Thespesia, Urena, In general, flavonols are represented by glycosides of quercetin, kaempferol, myricetin, herbacetin, gossypetin, and hibiscetin. However, flavonols and flavones with additional OH groups at the C-8 A ring and/or the C-5′ B ring positions are characteristic of this family, demonstrating chemotaxonomic significance. Key words: Flavones, flavonoids, flavonols, glycosides, Malvaceae, phytochemicals INTRODUCTION connate at least at their bases, but often forming a tube around the pistils. The pistils are composed of two to many connate he Malvaceae is a family of flowering carpels. The ovary is superior, with axial placentation, with plants estimated to contain 243 genera capitate or lobed stigma. The flowers have nectaries made with more than 4225 species. -

Total Phenolic Content, Free Radical Scavenging Capacity, and Anti- Cancer Activity of Silymarin
Journal of International Society for Food Bioactives Nutraceuticals and Functional Foods Review J. Food Bioact. 2020;10:53–63 Total phenolic content, free radical scavenging capacity, and anti- cancer activity of silymarin Uyory Choea, Monica Whenta*, Yinghua Luob and Liangli Yua aDepartment of Nutrition and Food Science, University of Maryland, College Park, MD 20742, USA bCollege of Food Science and Engineering, National Engineering Research Center for Fruit and Vegetable Processing, Ministry of Educa- tion, China Agricultural University, Beijing, China *Corresponding author: Monica Whent, Department of Nutrition and Food Science, University of Maryland, College Park, MD 20742, USA. Tel: +1 301 4054521; E-mail: [email protected] DOI: 10.31665/JFB.2020.10227 Received: June 19, 2020; Revised received & accepted: June 29, 2020 Citation: Choe, U., Whent, M., Luo, Y., and Yu, L. (2020). Total phenolic content, free radical scavenging capacity, and anti-cancer activity of silymarin. J. Food Bioact. 10: 53–63. Abstract Milk thistle (Silybum marianum) seeds are a good source of dietary polyphenols. The bioactive component of milk thistle seeds, silymarin, contains flavonolignans including silybin A, silybin B, isosilybin A, isosilybin B, sily- christin, isosilychristin, and silydiain along with the flavonol taxifolin. Silymarin is used traditionally as a natural herbal medicine with minimal side effects. Structurally, each silymarin component possesses phenolic hydroxyl groups and thus works as an antioxidant. In addition to free radical scavenging capacities, silymarin’s anti-cancer activities were reported for many different types of cancers including bladder, breast, colon, gastric, kidney, lung, oral, ovarian, prostate, and skin. The current review will discuss silymarin’s chemical components, total phenolic content, free radical scavenging capacities, and anti-cancer activities. -

Serbian Journal of Experimental and Clinical Research Vol12
Editor-in-Chief Slobodan Janković Co-Editors Nebojša Arsenijević, Miodrag Lukić, Miodrag Stojković, Milovan Matović, Slobodan Arsenijević, Nedeljko Manojlović, Vladimir Jakovljević, Mirjana Vukićević Board of Editors Ljiljana Vučković-Dekić, Institute for Oncology and Radiology of Serbia, Belgrade, Serbia Dragić Banković, Faculty for Natural Sciences and Mathematics, University of Kragujevac, Kragujevac, Serbia Zoran Stošić, Medical Faculty, University of Novi Sad, Novi Sad, Serbia Petar Vuleković, Medical Faculty, University of Novi Sad, Novi Sad, Serbia Philip Grammaticos, Professor Emeritus of Nuclear Medicine, Ermou 51, 546 23, Th essaloniki, Macedonia, Greece Stanislav Dubnička, Inst. of Physics Slovak Acad. Of Sci., Dubravska cesta 9, SK-84511 Bratislava, Slovak Republic Luca Rosi, SAC Istituto Superiore di Sanita, Vaile Regina Elena 299-00161 Roma, Italy Richard Gryglewski, Jagiellonian University, Department of Pharmacology, Krakow, Poland Lawrence Tierney, Jr, MD, VA Medical Center San Francisco, CA, USA Pravin J. Gupta, MD, D/9, Laxminagar, Nagpur – 440022 India Winfried Neuhuber, Medical Faculty, University of Erlangen, Nuremberg, Germany Editorial Staff Ivan Jovanović, Gordana Radosavljević, Nemanja Zdravković Vladislav Volarević Management Team Snezana Ivezic, Milan Milojevic, Bojana Radojevic, Ana Miloradovic Corrected by Scientifi c Editing Service “American Journal Experts” Design PrstJezikIostaliPsi - Miljan Nedeljković Print Medical Faculty, Kragujevac Indexed in EMBASE/Excerpta Medica, Index Copernicus, BioMedWorld, KoBSON, -
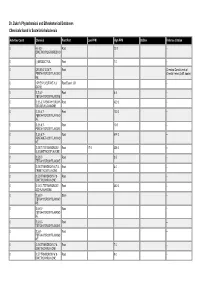
Dr. Duke's Phytochemical and Ethnobotanical Databases Chemicals Found in Scutellaria Baicalensis
Dr. Duke's Phytochemical and Ethnobotanical Databases Chemicals found in Scutellaria baicalensis Activities Count Chemical Plant Part Low PPM High PPM StdDev Refernce Citation 0 (+)-5,5'- Root 22.0 -- DIMETHOXYLARICIRESINO L 0 (-)-ERODICTYOL Root 7.0 -- 0 (2R,3R)-2',3,5,6',7- Root Chemical Constituents of PENTAHYDROXYFLAVONO Oriental Herbs (3 diff. books) NE 0 1-PHENYL-BUTANE-1,3- Root Essent. Oil -- DIONE 0 2',3',5,7- Root 6.4 -- TETRAHYDROXYFLAVONE 0 2',3,5,6',7-PENTAHYDROXY- Root 420.0 -- 2(R),3(R)-FLAVANONE 0 2',3,5,6',7- Root 120.0 -- PENTAHYDROXYFLAVANO NE 0 2',3,5,6',7- Root 10.0 -- PENTAHYDROXYFLAVONE 0 2',3,5,6',7- Root 674.0 -- PENTAMETHOXYFLAVANO NE 0 2',5,5',7-TETRAHYDROXY- Root 17.0 356.0 -- 6',8-DIMETHOXYFLAVONE 0 2',5,5',7- Root 3.5 -- TETRAHYDROXYFLAVONE 0 2',5,5'-TRIHYDROXY-6,7,8- Root 4.0 -- TRIMETHOXYFLAVONE 0 2',5,5'-TRIHYDROXY-7,8- Root -- DIMETHOXYFLAVONE 0 2',5,6',7-TETRAHYDROXY- Root 380.0 -- 2(S)-FLAVANONE 0 2',5,6',7- Stem -- TETRAHYDROXYFLAVANO NE 0 2',5,6',7- Root -- TETRAHYDROXYFLAVANO NE 0 2',5,6',7- Root -- TETRAHYDROXYFLAVONE 0 2',5,6'- Root -- TETRAHYDROXYFLAVANO NE 0 2',5,6'-TRIHYDROXY-7,8- Root 7.0 -- DIMETHOXYFLAVONE 0 2',5,7-TRIHYDROXY-6',8- Root 9.0 -- DIMETHOXYFLAVONE Activities Count Chemical Plant Part Low PPM High PPM StdDev Refernce Citation 0 2',5,7-TRIHYDROXY-6',8- Root 7.0 -- TRIMETHOXYFLAVONE 0 2',5,7-TRIHYDROXY-6'- Root 4.0 -- METHOXYFLAVONE 0 2',5,7-TRIHYDROXY-8- Root 6.0 -- METHOXYFLAVONE 0 2',5,7- Root 6.0 -- TRIHYDROXYFLAVONE 0 2',5,8-TRIHYDROXY-6,7- Root 200.0 -- DIMETHOXYFLAVONE 0 2'- Root -- METHYLSKULLCAPFLAVO NE 0 2(S),2',5,6',7- Root 230.0 -- TETRAHYDROXYFLAVANO NE 0 2,2',4',6-TETRAHYDROXY- Root 5.0 -- 6'-METHOXYCHALCONE 1 2,3-DIHYDROBENZOFURAN Root Essent. -

Flavonoids: Potential Candidates for the Treatment of Neurodegenerative Disorders
biomedicines Review Flavonoids: Potential Candidates for the Treatment of Neurodegenerative Disorders Shweta Devi 1,†, Vijay Kumar 2,*,† , Sandeep Kumar Singh 3,†, Ashish Kant Dubey 4 and Jong-Joo Kim 2,* 1 Systems Toxicology and Health Risk Assessment Group, CSIR-Indian Institute of Toxicology Research, Lucknow 226001, India; [email protected] 2 Department of Biotechnology, Yeungnam University, Gyeongsan, Gyeongbuk 38541, Korea 3 Department of Medical Genetics, SGPGIMS, Lucknow 226014, India; [email protected] 4 Department of Neurology, SGPGIMS, Lucknow 226014, India; [email protected] * Correspondence: [email protected] (V.K.); [email protected] (J.-J.K.); Tel.: +82-10-9668-3464 (J.-J.K.); Fax: +82-53-801-3464 (J.-J.K.) † These authors contributed equally to this work. Abstract: Neurodegenerative disorders, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD), are the most concerning disor- ders due to the lack of effective therapy and dramatic rise in affected cases. Although these disorders have diverse clinical manifestations, they all share a common cellular stress response. These cellular stress responses including neuroinflammation, oxidative stress, proteotoxicity, and endoplasmic reticulum (ER)-stress, which combats with stress conditions. Environmental stress/toxicity weakened the cellular stress response which results in cell damage. Small molecules, such as flavonoids, could reduce cellular stress and have gained much attention in recent years. Evidence has shown the poten- tial use of flavonoids in several ways, such as antioxidants, anti-inflammatory, and anti-apoptotic, yet their mechanism is still elusive. This review provides an insight into the potential role of flavonoids against cellular stress response that prevent the pathogenesis of neurodegenerative disorders. -

Expanding the Coverage of the Metabolic Landscape in Cultivated Rice with Integrated Computational Approaches
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.04.976266; this version posted March 5, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Expanding the Coverage of the Metabolic Landscape in 2 Cultivated Rice with Integrated Computational Approaches 3 4 5 Xuetong Li1,4,#,a, Hongxia Zhou1,4,#,b, Ning Xiao2,c, Xueting Wu1,d, Yuanhong Shan1,e, 6 Longxian Chen1, 4,f, Cuiting Wang1,g, Zixuan Wang1,h, Jirong Huang3,*,i, Aihong Li2,*,j, 7 and Xuan Li1,4,*,k 8 9 1Key Laboratory of Synthetic Biology, CAS Center for Excellence in Molecular Plant 10 Sciences, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, 11 Shanghai 200032, China 12 2Lixiahe Agricultural Research Institute of Jiangsu Province, Yangzhou 225007, 13 China 14 3Department of Biology, College of Life and Environmental Sciences, Shanghai 15 Normal University, Shanghai 200234, China 16 4University of Chinese Academy of Sciences, Beijing 100039, China 17 18 19 20 # Equal contribution. 21 * Corresponding authors. 22 Email: [email protected] (Li X), [email protected] (Li A), [email protected] 23 (Huang J) 24 25 26 Running title: Li X et al / Expanding the Coverage of the Metabolic Landscape 1 / 32 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.04.976266; this version posted March 5, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission.