In Transcriptional Regulation in Human Podocytes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alpha Actinin 4: an Intergral Component of Transcriptional
ALPHA ACTININ 4: AN INTERGRAL COMPONENT OF TRANSCRIPTIONAL PROGRAM REGULATED BY NUCLEAR HORMONE RECEPTORS By SIMRAN KHURANA Submitted in partial fulfillment of the requirements for the degree of doctor of philosophy Thesis Advisor: Dr. Hung-Ying Kao Department of Biochemistry CASE WESTERN RESERVE UNIVERSITY August, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of SIMRAN KHURANA ______________________________________________________ PhD candidate for the ________________________________degree *. Dr. David Samols (signed)_______________________________________________ (chair of the committee) Dr. Hung-Ying Kao ________________________________________________ Dr. Edward Stavnezer ________________________________________________ Dr. Leslie Bruggeman ________________________________________________ Dr. Colleen Croniger ________________________________________________ ________________________________________________ May 2011 (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS LIST OF TABLES vii LIST OF FIGURES viii ACKNOWLEDEMENTS xii LIST OF ABBREVIATIONS xiii ABSTRACT 1 CHAPTER 1: INTRODUCTION Family of Nuclear Receptors 3 Mechanism of transcriptional regulation by co-repressors and co-activators 8 Importance of LXXLL motif of co-activators in NR mediated transcription 12 Cyclic recruitment of co-regulators on the target promoters 15 Actin and actin related proteins (ABPs) in transcription -

List of Genes Associated with Sudden Cardiac Death (Scdgseta) Gene
List of genes associated with sudden cardiac death (SCDgseta) mRNA expression in normal human heart Entrez_I Gene symbol Gene name Uniprot ID Uniprot name fromb D GTEx BioGPS SAGE c d e ATP-binding cassette subfamily B ABCB1 P08183 MDR1_HUMAN 5243 √ √ member 1 ATP-binding cassette subfamily C ABCC9 O60706 ABCC9_HUMAN 10060 √ √ member 9 ACE Angiotensin I–converting enzyme P12821 ACE_HUMAN 1636 √ √ ACE2 Angiotensin I–converting enzyme 2 Q9BYF1 ACE2_HUMAN 59272 √ √ Acetylcholinesterase (Cartwright ACHE P22303 ACES_HUMAN 43 √ √ blood group) ACTC1 Actin, alpha, cardiac muscle 1 P68032 ACTC_HUMAN 70 √ √ ACTN2 Actinin alpha 2 P35609 ACTN2_HUMAN 88 √ √ √ ACTN4 Actinin alpha 4 O43707 ACTN4_HUMAN 81 √ √ √ ADRA2B Adrenoceptor alpha 2B P18089 ADA2B_HUMAN 151 √ √ AGT Angiotensinogen P01019 ANGT_HUMAN 183 √ √ √ AGTR1 Angiotensin II receptor type 1 P30556 AGTR1_HUMAN 185 √ √ AGTR2 Angiotensin II receptor type 2 P50052 AGTR2_HUMAN 186 √ √ AKAP9 A-kinase anchoring protein 9 Q99996 AKAP9_HUMAN 10142 √ √ √ ANK2/ANKB/ANKYRI Ankyrin 2 Q01484 ANK2_HUMAN 287 √ √ √ N B ANKRD1 Ankyrin repeat domain 1 Q15327 ANKR1_HUMAN 27063 √ √ √ ANKRD9 Ankyrin repeat domain 9 Q96BM1 ANKR9_HUMAN 122416 √ √ ARHGAP24 Rho GTPase–activating protein 24 Q8N264 RHG24_HUMAN 83478 √ √ ATPase Na+/K+–transporting ATP1B1 P05026 AT1B1_HUMAN 481 √ √ √ subunit beta 1 ATPase sarcoplasmic/endoplasmic ATP2A2 P16615 AT2A2_HUMAN 488 √ √ √ reticulum Ca2+ transporting 2 AZIN1 Antizyme inhibitor 1 O14977 AZIN1_HUMAN 51582 √ √ √ UDP-GlcNAc: betaGal B3GNT7 beta-1,3-N-acetylglucosaminyltransfe Q8NFL0 -

Alpha;-Actinin-4 Promotes Metastasis in Gastric Cancer
Laboratory Investigation (2017) 97, 1084–1094 © 2017 USCAP, Inc All rights reserved 0023-6837/17 α-Actinin-4 promotes metastasis in gastric cancer Xin Liu and Kent-Man Chu Metastasis increases the mortality rate of gastric cancer, which is the third leading cause of cancer-associated deaths worldwide. This study aims to identify the genes promoting metastasis of gastric cancer (GC). A human cell motility PCR array was used to analyze a pair of tumor and non-tumor tissue samples from a patient with stage IV GC (T3N3M1). Expression of the dysregulated genes was then evaluated in GC tissue samples (n = 10) and cell lines (n =6) via qPCR. Expression of α-actinin-4 (ACTN4) was validated in a larger sample size (n = 47) by qPCR, western blot and immunohistochemistry. Knockdown of ACTN4 with specific siRNAs was performed in GC cells, and adhesion assays, transwell invasion assays and migration assays were used to evaluate the function of these cells. Expression of potential targets of ACTN4 were then evaluated by qPCR. Thirty upregulated genes (greater than twofold) were revealed by the PCR array. We focused on ACTN4 because it was upregulated in 6 out of 10 pairs of tissue samples and 5 out of 6 GC cell lines. Further study indicated that ACTN4 was upregulated in 22/32 pairs of tissue samples at stage III & IV (P = 0.0069). Knockdown of ACTN4 in GC cells showed no significant effect on cell proliferation, but significantly increased cell-matrix adhesion, as well as reduced migration and invasion of AGS, MKN7 and NCI-N87 cells. -
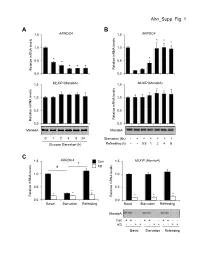
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

Proteomic Expression Profile in Human Temporomandibular Joint
diagnostics Article Proteomic Expression Profile in Human Temporomandibular Joint Dysfunction Andrea Duarte Doetzer 1,*, Roberto Hirochi Herai 1 , Marília Afonso Rabelo Buzalaf 2 and Paula Cristina Trevilatto 1 1 Graduate Program in Health Sciences, School of Medicine, Pontifícia Universidade Católica do Paraná (PUCPR), Curitiba 80215-901, Brazil; [email protected] (R.H.H.); [email protected] (P.C.T.) 2 Department of Biological Sciences, Bauru School of Dentistry, University of São Paulo, Bauru 17012-901, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-41-991-864-747 Abstract: Temporomandibular joint dysfunction (TMD) is a multifactorial condition that impairs human’s health and quality of life. Its etiology is still a challenge due to its complex development and the great number of different conditions it comprises. One of the most common forms of TMD is anterior disc displacement without reduction (DDWoR) and other TMDs with distinct origins are condylar hyperplasia (CH) and mandibular dislocation (MD). Thus, the aim of this study is to identify the protein expression profile of synovial fluid and the temporomandibular joint disc of patients diagnosed with DDWoR, CH and MD. Synovial fluid and a fraction of the temporomandibular joint disc were collected from nine patients diagnosed with DDWoR (n = 3), CH (n = 4) and MD (n = 2). Samples were subjected to label-free nLC-MS/MS for proteomic data extraction, and then bioinformatics analysis were conducted for protein identification and functional annotation. The three Citation: Doetzer, A.D.; Herai, R.H.; TMD conditions showed different protein expression profiles, and novel proteins were identified Buzalaf, M.A.R.; Trevilatto, P.C. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

The Biological Role of Actinin-4 (ACTN4)
Honda Cell Biosci (2015) 5:41 DOI 10.1186/s13578-015-0031-0 REVIEW Open Access The biological role of actinin‑4 (ACTN4) in malignant phenotypes of cancer Kazufumi Honda1,2* Abstract Invasion and metastasis are malignant phenotypes in cancer that lead to patient death. Cell motility is involved in these processes. In 1998, we identified overexpression of the actin-bundling protein actinin-4 in several types of can- cer. Protein expression of actinin-4 is closely associated with the invasive phenotypes of cancers. Actinin-4 is predomi- nantly expressed in the cellular protrusions that stimulate the invasive phenotype in cancer cells and is essential for formation of cellular protrusions such as filopodia and lamellipodia. ACTN4 (gene name encoding actinin-4 protein) is located on human chromosome 19q. ACTN4 amplification is frequently observed in patients with carcinomas of the pancreas, ovary, lung, and salivary gland, and patients with ACTN4 amplifications have worse outcomes than patients without amplification. In addition, nuclear distribution of actinin-4 is frequently observed in small cell lung, breast, and ovarian cancer. Actinin-4, when expressed in cancer cell nuclei, functions as a transcriptional co-activator. In this review, we summarize recent developments regarding the biological roles of actinin-4 in cancer invasion. Keywords: Cancer invasion, Metastasis, Actinin-4 (ACTN4), Actin-bundling protein, Biomarker Background Alpha-actinin is an actin cross-linking protein that Despite successful complete resection at the primary belongs to the spectrin superfamily. Four isoforms of cancer site, poor outcomes are occasionally observed alpha-actinin have been identified: alpha-actinin-1 (gene in patients due to failure to control distant metastasis. -

Functions of Nuclear Actin‑Binding Proteins in Human Cancer (Review)
ONCOLOGY LETTERS 15: 2743-2748, 2018 Functions of nuclear actin‑binding proteins in human cancer (Review) XINYI YANG1,2 and YING LIN1,2 1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation; 2Department of Gastroenterology and Hepatology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, Guangdong 510120, P.R. China Received February 16, 2017; Accepted November 10, 2017 DOI: 10.3892/ol.2017.7658 Abstract. Nuclear actin-binding proteins (ABPs) perform that contain actin-binding domains that interact with actin. distinguishable functions compared with their cytoplasmic They can bind to actin monomers, actin polymers or both (2). counterparts in extensive activities of living cells. In addition Early studies have focused on the biological features and to the ability to regulate actin cytoskeleton dynamics, nuclear physiological mechanisms of cytoplasmic actin. Therefore, ABPs are associated with multiple nuclear biological processes, ABPs were considered to be distributed only in the cytoplasm including chromatin remodeling, gene transcriptional regula- and associated with the organization of actin cytoskeleton (3). tion, DNA damage response, nucleocytoplasmic trafficking In the cytoplasm, actin is associated with numerous cellular and nuclear structure maintenance. The nuclear translocation activities, including sustaining cellular morphology, deter- of ABPs is affected by numerous intracellular or extracellular mining cellular organelle distribution, mediating intracellular stimuli, which may lead to developmental malformation, transfer, endocytosis and exocytosis, cell division, cell migration tumor initiation, tumor progression and metastasis. Abnormal and adhesion (4-8). Meanwhile, ABPs regulate actin cytoskel- expression of certain ABPs have been reported in different etal structure by modulating actin filament cross‑linking into types of cancer. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Characterization of Cardiac Progenitor Cell Activity in Engineered Heart Muscle
Characterization of cardiac progenitor cell activity in engineered heart muscle Doctoral Thesis In partial fulfillment of the requirements for the degree “Doctor rerum naturalium (Dr. rer. nat.)” in the Molecular Medicine Study Program at the Georg-August University Göttingen Submitted by Elif Levent born in Karabük, Turkey Göttingen 2016 Members of the Thesis Committee: Prof. Dr. med. Wolfram-Hubertus Zimmermann (Supervisor) Email: [email protected] Phone: +49 (0) 551 39 5781 Postal Address: Institute of Pharmacology and Toxicology University Medical Center Göttingen Georg-August University Göttingen Robert-Koch-Str. 40 37075 Göttingen Germany Prof. Dr. Dörthe Katschinski (Second member of thesis committee) Email: [email protected] Phone: +49 (0) 551 39 5896 Postal Address: Institute of Cardiovascular Physiology Humboldtallee 23 37073 Göttingen Germany Prof. Dr. Viacheslav O. Nikolaev (Third member of thesis committee) Email: [email protected] Phone: +49 (0) 40 7410 51391 Postal Address: Institute of Experimental Cardiovascular Research University Medical Center Hamburg-Eppendorf (UKE) Martinistr. 52 20246 Hamburg Date of disputation: 13.06.16 AFFIDAVIT Here I declare that my doctoral thesis entitled: “Characterization of cardiac progenitor cell activity in engineered heart muscle” has been written independently with no other sources and aids than quoted. Elif Levent Göttingen, April 2016 List of poster presentations Elif Levent, Malte Tiburcy, Amke Hesse, Dörthe Katschinski, Mark Sussman and Wolfram-H Zimmermann (2013). Hypoxia/Reoxygenated engineered heart muscle as an injury model system to investigate cardiac progenitor cell function in heart repair in vitro. Cardiorepair European multidisciplinary initiative (CARE-MI) annual meeting Lisbon, Portugal. Elif Levent, Malte Tiburcy, Mark Sussman and Wolfram-H Zimmermann (2013). -
![RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)](https://docslib.b-cdn.net/cover/3271/rt%C2%B2-profiler-pcr-array-96-well-format-and-384-well-4-x-96-format-3353271.webp)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 x 96] Format) Mouse Focal Adhesions Cat. no. 330231 PAMM-145ZA For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Applied Biosystems® models 5700, 7000, 7300, 7500, Format A 7700, 7900HT, ViiA™ 7 (96-well block); Bio-Rad® models iCycler®, iQ™5, MyiQ™, MyiQ2; Bio-Rad/MJ Research Chromo4™; Eppendorf® Mastercycler® ep realplex models 2, 2s, 4, 4s; Stratagene® models Mx3005P®, Mx3000P®; Takara TP-800 RT² Profiler PCR Array, Applied Biosystems models 7500 (Fast block), 7900HT (Fast Format C block), StepOnePlus™, ViiA 7 (Fast block) RT² Profiler PCR Array, Bio-Rad CFX96™; Bio-Rad/MJ Research models DNA Format D Engine Opticon®, DNA Engine Opticon 2; Stratagene Mx4000® RT² Profiler PCR Array, Applied Biosystems models 7900HT (384-well block), ViiA 7 Format E (384-well block); Bio-Rad CFX384™ RT² Profiler PCR Array, Roche® LightCycler® 480 (96-well block) Format F RT² Profiler PCR Array, Roche LightCycler 480 (384-well block) Format G RT² Profiler PCR Array, Fluidigm® BioMark™ Format H Sample & Assay Technologies Description The Mouse Focal Adhesions RT² Profiler PCR Array profiles the expression of 84 key genes involved in cellular adhesion to the extracellular matrix (ECM). Focal adhesions and hemidesmosomes form around the intracellular domains of integrins bound to components of the ECM such as those found in the basal lamina underlying epithelial cells. Focal adhesions connect the intracellular domains of integrins to actin filaments, while hemidesmosomes connect them to keratin-based intermediate filaments. These structures regulate several key normal biological processes including angiogenesis, anchorage-dependent cell survival, cell cycle, cell migration, and wound healing. -

Cancer Testis Antigen Promotes Triple Negative Breast Cancer
www.nature.com/scientificreports Corrected: Author Correction OPEN Cancer Testis Antigen Promotes Triple Negative Breast Cancer Metastasis and is Traceable in the Received: 31 January 2019 Accepted: 16 July 2019 Circulating Extracellular Vesicles Published online: 12 August 2019 Anbarasu Kannan1, Julie V. Philley2, Kate L. Hertweck3,8, Harrison Ndetan4, Karan P. Singh4, Subramaniam Sivakumar5, Robert B. Wells6, Ratna K. Vadlamudi 7 & Santanu Dasgupta 1,2 Triple negative breast cancer (TNBC) has poor survival, exhibits rapid metastases, lacks targeted therapies and reliable prognostic markers. Here, we examined metastasis promoting role of cancer testis antigen SPANXB1 in TNBC and its utility as a therapeutic target and prognostic biomarker. Expression pattern of SPANXB1 was determined using matched primary cancer, lymph node metastatic tissues and circulating small extracellular vesicles (sEVs). cDNA microarray analysis of TNBC cells stably integrated with a metastasis suppressor SH3GL2 identifed SPANXB1 as a potential target gene. TNBC cells overexpressing SH3GL2 exhibited decreased levels of both SPANXB1 mRNA and protein. Silencing of SPANXB1 reduced migration, invasion and reactive oxygen species production of TNBC cells. SPANXB1 depletion augmented SH3GL2 expression and decreased RAC-1, FAK, A-Actinin and Vinculin expression. Phenotypic and molecular changes were reversed upon SPANXB1 re-expression. SPANXB1 overexpressing breast cancer cells with an enhanced SPANXB1:SH3GL2 ratio achieved pulmonary metastasis within 5 weeks, whereas controls cells failed to do so. Altered expression of SPANXB1 was detected in the sEVs of SPANXB1 transduced cells. Exclusive expression of SPANXB1 was traceable in circulating sEVs, which was associated with TNBC progression. SPANXB1 represents a novel and ideal therapeutic target for blocking TNBC metastases due to its unique expression pattern and may function as an EV based prognostic marker to improve TNBC survival.