Characterization of Cardiac Progenitor Cell Activity in Engineered Heart Muscle
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alpha Actinin 4: an Intergral Component of Transcriptional
ALPHA ACTININ 4: AN INTERGRAL COMPONENT OF TRANSCRIPTIONAL PROGRAM REGULATED BY NUCLEAR HORMONE RECEPTORS By SIMRAN KHURANA Submitted in partial fulfillment of the requirements for the degree of doctor of philosophy Thesis Advisor: Dr. Hung-Ying Kao Department of Biochemistry CASE WESTERN RESERVE UNIVERSITY August, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of SIMRAN KHURANA ______________________________________________________ PhD candidate for the ________________________________degree *. Dr. David Samols (signed)_______________________________________________ (chair of the committee) Dr. Hung-Ying Kao ________________________________________________ Dr. Edward Stavnezer ________________________________________________ Dr. Leslie Bruggeman ________________________________________________ Dr. Colleen Croniger ________________________________________________ ________________________________________________ May 2011 (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS LIST OF TABLES vii LIST OF FIGURES viii ACKNOWLEDEMENTS xii LIST OF ABBREVIATIONS xiii ABSTRACT 1 CHAPTER 1: INTRODUCTION Family of Nuclear Receptors 3 Mechanism of transcriptional regulation by co-repressors and co-activators 8 Importance of LXXLL motif of co-activators in NR mediated transcription 12 Cyclic recruitment of co-regulators on the target promoters 15 Actin and actin related proteins (ABPs) in transcription -

List of Genes Associated with Sudden Cardiac Death (Scdgseta) Gene
List of genes associated with sudden cardiac death (SCDgseta) mRNA expression in normal human heart Entrez_I Gene symbol Gene name Uniprot ID Uniprot name fromb D GTEx BioGPS SAGE c d e ATP-binding cassette subfamily B ABCB1 P08183 MDR1_HUMAN 5243 √ √ member 1 ATP-binding cassette subfamily C ABCC9 O60706 ABCC9_HUMAN 10060 √ √ member 9 ACE Angiotensin I–converting enzyme P12821 ACE_HUMAN 1636 √ √ ACE2 Angiotensin I–converting enzyme 2 Q9BYF1 ACE2_HUMAN 59272 √ √ Acetylcholinesterase (Cartwright ACHE P22303 ACES_HUMAN 43 √ √ blood group) ACTC1 Actin, alpha, cardiac muscle 1 P68032 ACTC_HUMAN 70 √ √ ACTN2 Actinin alpha 2 P35609 ACTN2_HUMAN 88 √ √ √ ACTN4 Actinin alpha 4 O43707 ACTN4_HUMAN 81 √ √ √ ADRA2B Adrenoceptor alpha 2B P18089 ADA2B_HUMAN 151 √ √ AGT Angiotensinogen P01019 ANGT_HUMAN 183 √ √ √ AGTR1 Angiotensin II receptor type 1 P30556 AGTR1_HUMAN 185 √ √ AGTR2 Angiotensin II receptor type 2 P50052 AGTR2_HUMAN 186 √ √ AKAP9 A-kinase anchoring protein 9 Q99996 AKAP9_HUMAN 10142 √ √ √ ANK2/ANKB/ANKYRI Ankyrin 2 Q01484 ANK2_HUMAN 287 √ √ √ N B ANKRD1 Ankyrin repeat domain 1 Q15327 ANKR1_HUMAN 27063 √ √ √ ANKRD9 Ankyrin repeat domain 9 Q96BM1 ANKR9_HUMAN 122416 √ √ ARHGAP24 Rho GTPase–activating protein 24 Q8N264 RHG24_HUMAN 83478 √ √ ATPase Na+/K+–transporting ATP1B1 P05026 AT1B1_HUMAN 481 √ √ √ subunit beta 1 ATPase sarcoplasmic/endoplasmic ATP2A2 P16615 AT2A2_HUMAN 488 √ √ √ reticulum Ca2+ transporting 2 AZIN1 Antizyme inhibitor 1 O14977 AZIN1_HUMAN 51582 √ √ √ UDP-GlcNAc: betaGal B3GNT7 beta-1,3-N-acetylglucosaminyltransfe Q8NFL0 -

On the Turning of Xenopus Retinal Axons Induced by Ephrin-A5
Development 130, 1635-1643 1635 © 2003 The Company of Biologists Ltd doi:10.1242/dev.00386 On the turning of Xenopus retinal axons induced by ephrin-A5 Christine Weinl1, Uwe Drescher2,*, Susanne Lang1, Friedrich Bonhoeffer1 and Jürgen Löschinger1 1Max-Planck-Institute for Developmental Biology, Spemannstrasse 35, 72076 Tübingen, Germany 2MRC Centre for Developmental Neurobiology, King’s College London, New Hunt’s House, Guy’s Hospital Campus, London SE1 1UL, UK *Author for correspondence (e-mail: [email protected]) Accepted 13 January 2003 SUMMARY The Eph family of receptor tyrosine kinases and their turning or growth cone collapse when confronted with ligands, the ephrins, play important roles during ephrin-A5-Fc bound to beads. However, when added in development of the nervous system. Frequently they exert soluble form to the medium, ephrin-A5 induces growth their functions through a repellent mechanism, so that, for cone collapse, comparable to data from chick. example, an axon expressing an Eph receptor does not The analysis of growth cone behaviour in a gradient of invade a territory in which an ephrin is expressed. Eph soluble ephrin-A5 in the ‘turning assay’ revealed a receptor activation requires membrane-associated ligands. substratum-dependent reaction of Xenopus retinal axons. This feature discriminates ephrins from other molecules On fibronectin, we observed a repulsive response, with the sculpturing the nervous system such as netrins, slits and turning of growth cones away from higher concentrations class 3 semaphorins, which are secreted molecules. While of ephrin-A5. On laminin, retinal axons turned towards the ability of secreted molecules to guide axons, i.e. -

Endothelin-2 Signaling in the Neural Retina Promotes the Endothelial Tip Cell State and Inhibits Angiogenesis
Endothelin-2 signaling in the neural retina promotes PNAS PLUS the endothelial tip cell state and inhibits angiogenesis Amir Rattnera,1, Huimin Yua, John Williamsa,b, Philip M. Smallwooda,b, and Jeremy Nathansa,b,c,d,1 Departments of aMolecular Biology and Genetics, cNeuroscience, and dOphthalmology and bHoward Hughes Medical Institute, Johns Hopkins University School of Medicine, Baltimore, MD 21205 Contributed by Jeremy Nathans, August 20, 2013 (sent for review February 19, 2013) Endothelin signaling is required for neural crest migration and time lapse imaging studies of vascular development in zebrafish homeostatic regulation of blood pressure. Here, we report that and mammalian EC dynamics in explant culture show that the tip constitutive overexpression of Endothelin-2 (Edn2) in the mouse cell and stalk cell states are highly plastic, with frequent retina perturbs vascular development by inhibiting endothelial cell exchanges between the two cell states (8, 9). migration across the retinal surface and subsequent endothelial Several other signaling pathways are also essential for retinal cell invasion into the retina. Developing endothelial cells exist in vascular development. Norrin, a Muller-glia–derived ligand, and one of two states: tip cells at the growing front and stalk cells in its EC receptor Frizzled4 (Fz4), coreceptor Lrp5, and receptor the vascular plexus behind the front. This division of endothelial chaperone Tspan12 activate canonical Wnt signaling in de- cell states is one of the central organizing principles of angiogen- veloping ECs (10). In humans and mice, defects in any of these esis. In the developing retina, Edn2 overexpression leads to components lead to retinal hypovascularization. -

Mir-146A Regulates Migration and Invasion by Targeting NRP2 in Circulating-Tumor Cell Mimicking Suspension Cells
G C A T T A C G G C A T genes Article MiR-146a Regulates Migration and Invasion by Targeting NRP2 in Circulating-Tumor Cell Mimicking Suspension Cells Yeojin Do 1, Jin Gu Cho 1, Ji Young Park 1, Sumin Oh 1, Doyeon Park 1 , Kyung Hyun Yoo 1,2 , Myeong-Sok Lee 1,2, Byung Su Kwon 3,* , Jongmin Kim 1,2,* and Young Yang 2,* 1 Division of Biological Sciences, Sookmyung Women’s University, Seoul 04310, Korea; [email protected] (Y.D.); [email protected] (J.G.C.); [email protected] (J.Y.P.); [email protected] (S.O.); [email protected] (D.P.); [email protected] (K.H.Y.); [email protected] (M.-S.L.) 2 Research Institute for Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea 3 Department of Obstetrics and Gynecology, Kyung Hee University Medical Center, 23, Seoul 02447, Korea * Correspondence: [email protected] (B.S.K.); [email protected] (J.K.); [email protected] (Y.Y.); Tel.: +82-2-958-8837 (B.S.K.); +82-2-710-9553 (J.K.); +82-2-710-9590 (Y.Y.) Abstract: Cancer metastasis is the primary cause of cancer-related death and metastatic cancer has circulating-tumor cells (CTCs), which circulate in the bloodstream before invading other organs. Thus, understanding the precise role of CTCs may provide new insights into the metastasis process and reduce cancer mortality. However, the molecular characteristics of CTCs are not well understood due to a lack of number of CTCs. -

Alpha;-Actinin-4 Promotes Metastasis in Gastric Cancer
Laboratory Investigation (2017) 97, 1084–1094 © 2017 USCAP, Inc All rights reserved 0023-6837/17 α-Actinin-4 promotes metastasis in gastric cancer Xin Liu and Kent-Man Chu Metastasis increases the mortality rate of gastric cancer, which is the third leading cause of cancer-associated deaths worldwide. This study aims to identify the genes promoting metastasis of gastric cancer (GC). A human cell motility PCR array was used to analyze a pair of tumor and non-tumor tissue samples from a patient with stage IV GC (T3N3M1). Expression of the dysregulated genes was then evaluated in GC tissue samples (n = 10) and cell lines (n =6) via qPCR. Expression of α-actinin-4 (ACTN4) was validated in a larger sample size (n = 47) by qPCR, western blot and immunohistochemistry. Knockdown of ACTN4 with specific siRNAs was performed in GC cells, and adhesion assays, transwell invasion assays and migration assays were used to evaluate the function of these cells. Expression of potential targets of ACTN4 were then evaluated by qPCR. Thirty upregulated genes (greater than twofold) were revealed by the PCR array. We focused on ACTN4 because it was upregulated in 6 out of 10 pairs of tissue samples and 5 out of 6 GC cell lines. Further study indicated that ACTN4 was upregulated in 22/32 pairs of tissue samples at stage III & IV (P = 0.0069). Knockdown of ACTN4 in GC cells showed no significant effect on cell proliferation, but significantly increased cell-matrix adhesion, as well as reduced migration and invasion of AGS, MKN7 and NCI-N87 cells. -
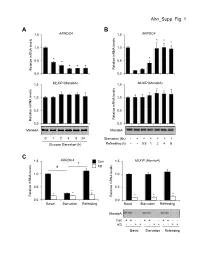
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

3B and Semaphorin-3F in Ovarian Cancer
Published OnlineFirst February 2, 2010; DOI: 10.1158/1535-7163.MCT-09-0664 Research Article Molecular Cancer Therapeutics Hormonal Regulation and Distinct Functions of Semaphorin- 3B and Semaphorin-3F in Ovarian Cancer Doina Joseph1, Shuk-Mei Ho2, and Viqar Syed1 Abstract Semaphorins comprise a family of molecules that influence neuronal growth and guidance. Class-3 sema- phorins, semaphorin-3B (SEMA3B) and semaphorin-3F (SEMA3F), illustrate their effects by forming a com- plex with neuropilins (NP-1 or NP-2) and plexins. We examined the status and regulation of semaphorins and their receptors in human ovarian cancer cells. A significantly reduced expression of SEMA3B (83 kDa), SE- MA3F (90 kDa), and plexin-A3 was observed in ovarian cancer cell lines when compared with normal human ovarian surface epithelial cells. The expression of NP-1, NP-2, and plexin-A1 was not altered in human ovar- ian surface epithelial and ovarian cancer cells. The decreased expression of SEMA3B, SEMA3F, and plexin-A3 was confirmed in stage 3 ovarian tumors. The treatment of ovarian cancer cells with luteinizing hormone, follicle-stimulating hormone, and estrogen induced a significant upregulation of SEMA3B, whereas SEMA3F was upregulated only by estrogen. Cotreatment of cell lines with a hormone and its specific antagonist blocked the effect of the hormone. Ectopic expression of SEMA3B or SEMA3F reduced soft-agar colony for- mation, adhesion, and cell invasion of ovarian cancer cell cultures. Forced expression of SEMA3B, but not SEMA3F, inhibited viability of ovarian cancer cells. Overexpression of SEMA3B and SEMA3F reduced focal adhesion kinase phosphorylation and matrix metalloproteinase-2 and matrix metalloproteinase-9 expression in ovarian cancer cells. -

Proteomic Expression Profile in Human Temporomandibular Joint
diagnostics Article Proteomic Expression Profile in Human Temporomandibular Joint Dysfunction Andrea Duarte Doetzer 1,*, Roberto Hirochi Herai 1 , Marília Afonso Rabelo Buzalaf 2 and Paula Cristina Trevilatto 1 1 Graduate Program in Health Sciences, School of Medicine, Pontifícia Universidade Católica do Paraná (PUCPR), Curitiba 80215-901, Brazil; [email protected] (R.H.H.); [email protected] (P.C.T.) 2 Department of Biological Sciences, Bauru School of Dentistry, University of São Paulo, Bauru 17012-901, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-41-991-864-747 Abstract: Temporomandibular joint dysfunction (TMD) is a multifactorial condition that impairs human’s health and quality of life. Its etiology is still a challenge due to its complex development and the great number of different conditions it comprises. One of the most common forms of TMD is anterior disc displacement without reduction (DDWoR) and other TMDs with distinct origins are condylar hyperplasia (CH) and mandibular dislocation (MD). Thus, the aim of this study is to identify the protein expression profile of synovial fluid and the temporomandibular joint disc of patients diagnosed with DDWoR, CH and MD. Synovial fluid and a fraction of the temporomandibular joint disc were collected from nine patients diagnosed with DDWoR (n = 3), CH (n = 4) and MD (n = 2). Samples were subjected to label-free nLC-MS/MS for proteomic data extraction, and then bioinformatics analysis were conducted for protein identification and functional annotation. The three Citation: Doetzer, A.D.; Herai, R.H.; TMD conditions showed different protein expression profiles, and novel proteins were identified Buzalaf, M.A.R.; Trevilatto, P.C. -

Semaphorin Signaling Guides Cranial Neural Crest Cell Migration in Zebrafish
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Developmental Biology 280 (2005) 373–385 www.elsevier.com/locate/ydbio Semaphorin signaling guides cranial neural crest cell migration in zebrafish Hung-Hsiang Yu, Cecilia B. Moens* Howard Hughes Medical Institute and Division of Basic Science, Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue N., Box 19024 Seattle, WA 98109, USA Received for publication 2 December 2004, revised 19 January 2005, accepted 28 January 2005 Abstract Cranial neural crest cells (NCCs) migrate into the pharyngeal arches in three primary streams separated by two cranial neural crest (NC)- free zones. Multiple tissues have been implicated in the guidance of cranial NCC migration; however, the signals provided by these tissues have remained elusive. We investigate the function of semaphorins (semas) and their receptors, neuropilins (nrps), in cranial NCC migration in zebrafish. We find that genes of the sema3F and sema3G class are expressed in the cranial NC-free zones, while nrp2a and nrp2b are expressed in the migrating NCCs. sema3F/3G expression is expanded homogeneously in the head periphery through which the cranial NCCs migrate in lzr/pbx4 mutants, in which the cranial NC streams are fused. Antisense morpholino knockdown of Sema3F/3G or Nrp2 suppresses the abnormal cranial NC phenotype of lzr/pbx4 mutants, demonstrating that aberrant Sema3F/3G-Nrp2 signaling is responsible for this phenotype and suggesting that repulsive Sema3F/3G-Npn2 signaling normally contributes to the guidance of migrating cranial NCCs. Furthermore, global over-expression of sema3Gb phenocopies the aberrant cranial NC phenotype of lzr/pbx4 mutants when endogenous Sema3 ligands are knocked down, consistent with a model in which the patterned expression of Sema3 ligands in the head periphery coordinates the migration of Nrp-expressing cranial NCCs. -

Hepatocyte Growth Factor: a Regulator of Inflammation and Autoimmunity
Autoimmunity Reviews 14 (2015) 293–303 Contents lists available at ScienceDirect Autoimmunity Reviews journal homepage: www.elsevier.com/locate/autrev Review Hepatocyte growth factor: A regulator of inflammation and autoimmunity Nicolas Molnarfi a,b,1, Mahdia Benkhoucha a,b,1, Hiroshi Funakoshi d, Toshikazu Nakamura e, Patrice H. Lalive a,b,c,⁎ a Department of Pathology and Immunology, Faculty of Medicine, University of Geneva, Geneva, Switzerland b Department of Clinical Neurosciences, Division of Neurology, Unit of Neuroimmunology and Multiple Sclerosis, University Hospital of Geneva, Geneva, Switzerland c Department of Genetics and Laboratory Medicine, Laboratory Medicine Service, University Hospital of Geneva, Geneva, Switzerland d Center for Advanced Research and Education, Asahikawa Medical University, Asahikawa, Japan e Neurogen Inc., Nakahozumi, Ibaraki, Osaka, Japan article info abstract Article history: Hepatocyte growth factor (HGF) is a pleiotropic cytokine that has been extensively studied over several decades, Received 20 November 2014 but was only recently recognized as a key player in mediating protection of many types of inflammatory and au- Accepted 25 November 2014 toimmune diseases. HGF was reported to prevent and attenuate disease progression by influencing multiple Available online 1 December 2014 pathophysiological processes involved in inflammatory and immune response, including cell migration, matura- tion, cytokine production, antigen presentation, and T cell effector function. In this review, we discuss the actions Keywords: fl HGF and mechanisms of HGF in in ammation and immunity and the therapeutic potential of this factor for the treat- fl c-Met ment of in ammatory and autoimmune diseases. Inflammation © 2014 Elsevier B.V. All rights reserved. Autoimmunity Autoimmune regulator Therapy Contents 1. -

1,25-Dihydroxyvitamin D3-Induced Genes in Osteoblasts
1,25-DIHYDROXYVITAMIN D3-INDUCED GENES IN OSTEOBLASTS: UNCOVERING NEW FUNCTIONS FOR MENINGIOMA 1 AND SEMAPHORIN 3B IN SKELETAL PHYSIOLOGY by XIAOXUE ZHANG Submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Thesis advisor: Paul N. MacDonald Department of Pharmacology CASE WESTERN RESERVE UNIVERSITY May 2009 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of _____________________________________________________ candidate for the ______________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. I dedicate this thesis to my mother and father for their lifelong love, encouragement and sacrifice TABLE OF CONTENTS Table of Contents ii List of Tables iii List of Figures iv Acknowledgements vii Abbreviations x Abstract xiii Chapter I Introduction 1 Chapter II Meningioma 1 (MN1) is a 1,25-dihydroxyvitamin D3- 44 induced transcription coactivator that promotes osteoblast proliferation, motility, differentiation, and function Chapter III Semaphorin 3B (SEMA3B) is a 1,25- 108 dihydroxyvitamin D3-induced gene in osteoblasts that promotes