And Exo1 Interaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconstitution of Long and Short Patch Mismatch Repair Reactions Using Saccharomyces Cerevisiae Proteins
Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins Nikki Bowena, Catherine E. Smitha, Anjana Srivatsana, Smaranda Willcoxb,c, Jack D. Griffithb,c, and Richard D. Kolodnera,d,e,f,g,1 aLudwig Institute for Cancer Research, Departments of dMedicine and eCellular and Molecular Medicine, fMoores-University of California, San Diego Cancer Center, and gInstitute of Genomic Medicine, University of California San Diego School of Medicine, La Jolla, CA 92093; and bLineberger Cancer Center and cDepartment of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27514 Contributed by Richard D. Kolodner, October 8, 2013 (sent for review September 9, 2013) A problem in understanding eukaryotic DNA mismatch repair In eukaryotic MMR, mispairs are bound by MutS homolog 2 (MMR) mechanisms is linking insights into MMR mechanisms from (Msh2)–MutS homolog 6 (Msh6) and Msh2–MutS homolog 3 genetics and cell-biology studies with those from biochemical (Msh3), two partially redundant complexes of MutS-related pro- studies of MMR proteins and reconstituted MMR reactions. This teins (3, 4, 18, 19). These complexes recruit a MutL-related type of analysis has proven difficult because reconstitution ap- complex, called MutL homoloh 1 (Mlh1)–postmeiotic segrega- proaches have been most successful for human MMR whereas tion 1 (Pms1) in S. cerevisiae and Mlh1–postmeiotic segregation – – analysis of MMR in vivo has been most advanced in the yeast 2 (Pms2) in human and mouse (3, 4, 20 23). The Mlh1 Pms1/ Saccharomyces cerevisiae. Here, we describe the reconstitution of Pms2 complex has an endonuclease activity suggested to play MMR reactions using purified S. -

Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA
Mechanisms in E. coli and Human Mismatch Repair Nobel Lecture, December 8, 2015 by Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA. he idea that mismatched base pairs occur in cells and that such lesions trig- T ger their own repair was suggested 50 years ago by Robin Holliday in the context of genetic recombination [1]. Breakage and rejoining of DNA helices was known to occur during this process [2], with precision of rejoining attributed to formation of a heteroduplex joint, a region of helix where the two strands are derived from the diferent recombining partners. Holliday pointed out that if this heteroduplex region should span a genetic diference between the two DNAs, then it will contain one or more mismatched base pairs. He invoked processing of such mismatches to explain the recombination-associated phenomenon of gene conversion [1], noting that “If there are enzymes which can repair points of damage in DNA, it would seem possible that the same enzymes could recognize the abnormality of base pairing, and by exchange reactions rectify this.” Direct evidence that mismatches provoke a repair reaction was provided by bacterial transformation experiments [3–5], and our interest in this efect was prompted by the Escherichia coli (E. coli) work done in Matt Meselson’s lab at Harvard. Using artifcially constructed heteroduplex DNAs containing multiple mismatched base pairs, Wagner and Meselson [6] demonstrated that mismatches elicit a repair reaction upon introduction into the E. coli cell. Tey also showed that closely spaced mismatches, mismatches separated by a 1000 base pairs or so, are usually repaired on the same DNA strand. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Phosphate Steering by Flap Endonuclease 1 Promotes 50-flap Specificity and Incision to Prevent Genome Instability
ARTICLE Received 18 Jan 2017 | Accepted 5 May 2017 | Published 27 Jun 2017 DOI: 10.1038/ncomms15855 OPEN Phosphate steering by Flap Endonuclease 1 promotes 50-flap specificity and incision to prevent genome instability Susan E. Tsutakawa1,*, Mark J. Thompson2,*, Andrew S. Arvai3,*, Alexander J. Neil4,*, Steven J. Shaw2, Sana I. Algasaier2, Jane C. Kim4, L. David Finger2, Emma Jardine2, Victoria J.B. Gotham2, Altaf H. Sarker5, Mai Z. Her1, Fahad Rashid6, Samir M. Hamdan6, Sergei M. Mirkin4, Jane A. Grasby2 & John A. Tainer1,7 DNA replication and repair enzyme Flap Endonuclease 1 (FEN1) is vital for genome integrity, and FEN1 mutations arise in multiple cancers. FEN1 precisely cleaves single-stranded (ss) 50-flaps one nucleotide into duplex (ds) DNA. Yet, how FEN1 selects for but does not incise the ss 50-flap was enigmatic. Here we combine crystallographic, biochemical and genetic analyses to show that two dsDNA binding sites set the 50polarity and to reveal unexpected control of the DNA phosphodiester backbone by electrostatic interactions. Via ‘phosphate steering’, basic residues energetically steer an inverted ss 50-flap through a gateway over FEN1’s active site and shift dsDNA for catalysis. Mutations of these residues cause an 18,000-fold reduction in catalytic rate in vitro and large-scale trinucleotide (GAA)n repeat expansions in vivo, implying failed phosphate-steering promotes an unanticipated lagging-strand template-switch mechanism during replication. Thus, phosphate steering is an unappreciated FEN1 function that enforces 50-flap specificity and catalysis, preventing genomic instability. 1 Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. -
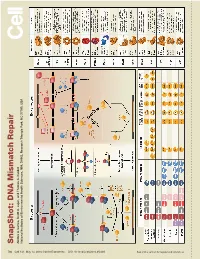
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible -

Download (PDF)
Supplemental Table 1A Fold change Affymetrix probe Gene title Gene symbol (log2 ratio to ID control) 1424525_at gastrin releasing peptide Grp 5.80 1427747_a_at lipocalin 2 Lcn2 5.27 1449833_at small proline‐rich protein 2F Sprr2f 5.04 1450297_at interleukin 6 Il6 5.02 1450364_a_at hepatitis A virus cellular receptor 1 Havcr1 4.42 1452098_at CTF18, chromosome transmission fidelity factor 18 homolog (S. cerevisiae) Chtf18 4.25 1419282_at chemokine (C‐C motif) ligand 12 Ccl12 4.11 serine (or cysteine) peptidase inhibitor, clade A (alpha‐1 antiproteinase, 1424758_s_at Serpina10 3.96 antitrypsin), member 10 1460227_at tissue inhibitor of metalloproteinase 1 Timp1 3.86 1419100_at serine (or cysteine) peptidase inhibitor, clade A, member 3N Serpina3n 3.59 1418626_a_at clusterin Clu 3.31 1416025_at fibrinogen, gamma polypeptide Fgg 3.25 1448756_at S100 calcium binding protein A9 (calgranulin B) S100a9 3.18 1427682_a_at early growth response 2 Egr2 3.17 1452279_at complement factor properdin Cfp 3.03 1422851_at high mobility group AT‐hook 2 Hmga2 3.01 1419579_at solute carrier family 7 (cationic amino acid transporter, y+ system), member 12 Slc7a12 2.98 1419209_at chemokine (C‐X‐C motif) ligand 1 Cxcl1 2.92 1419764_at chitinase 3‐like 3 Chi3l3 2.91 1423954_at complement component 3 C3 2.88 1424279_at fibrinogen, alpha polypeptide Fga 2.86 1424556_at pyrroline‐5‐carboxylate reductase 1 Pycr1 2.77 1418778_at coiled‐coil domain containing 109B Ccdc109b 2.76 1422864_at runt related transcription factor 1 Runx1 2.65 1433966_x_at asparagine synthetase -

Three Prime Repair Exonuclease 1 (TREX1) Expression Correlates with Cervical Cancer Cells Growth in Vitro and Disease Progressio
www.nature.com/scientificreports OPEN Three Prime Repair Exonuclease 1 (TREX1) expression correlates with cervical cancer cells growth in vitro Received: 11 December 2017 Accepted: 17 October 2018 and disease progression in vivo Published: xx xx xxxx Bruna Prati1, Walason da Silva Abjaude1, Lara Termini2, Mirian Morale3, Suellen Herbster1, Adhemar Longatto-Filho4,5,6, Rafaella Almeida Lima Nunes2, Lizeth Carolina Córdoba Camacho1,11,12, Silvia Helena Rabelo-Santos7, Luiz Carlos Zeferino 8, Francisco Aguayo9,10 & Enrique Boccardo 1 Alterations in specifc DNA damage repair mechanisms in the presence of human papillomavirus (HPV) infection have been described in diferent experimental models. However, the global efect of HPV on the expression of genes involved in these pathways has not been analyzed in detail. In the present study, we compared the expression profle of 135 genes involved in DNA damage repair among primary human keratinocytes (PHK), HPV-positive (SiHa and HeLa) and HPV-negative (C33A) cervical cancer derived cell lines. We identifed 9 genes which expression pattern distinguishes HPV-positive tumor cell lines from C33A. Moreover, we observed that Three Prime Repair Exonuclease 1 (TREX1) expression is upregulated exclusively in HPV-transformed cell lines and PHK expressing HPV16 E6 and E7 oncogenes. We demonstrated that TREX1 silencing greatly afects tumor cells clonogenic and anchorage independent growth potential. We showed that this efect is associated with p53 upregulation, accumulation of subG1 cells, and requires the expression of E7 from high-risk HPV types. Finally, we observed an increase in TREX1 levels in precancerous lesions, squamous carcinomas and adenocarcinomas clinical samples. Altogether, our results indicate that TREX1 upregulation is important for cervical tumor cells growth and may contribute with tumor establishment and progression. -

The Devil Is in the Details for DNA Mismatch Repair COMMENTARY
COMMENTARY The Devil is in the details for DNA mismatch repair COMMENTARY Peggy Hsieha,1 and Yongliang Zhanga Ensuring high-fidelity DNA replication is essential a strand-specific signal for excision can be far apart) (7–9). for maintaining genome stability in organisms from MMR in eukaryotes features two families of MMR pro- Escherichia coli to humans. This task requires an intricate teins, heterodimeric homologs of bacterial MutS (MSH) network of cellular components that carries out replica- or MutL (MLH). Early models of E. coli MMR invoke tion and postreplication DNA repair (1). In eukaryotes, MMR protein-mediated DNA looping between a mis- replication is carried out by Pol e and Pol δ that synthesize match and a DNA-methylation excision signal or ATP- the leading and lagging strands, respectively, using short driven MutS translocation along the DNA. The nucleotide RNA–DNA primers synthesized by Pol α. Although repli- switch model posits the existence of multiple diffusing cation gives rise to a low spontaneous mutation rate of MSH–MLH clamps (8). Other studies suggest that a single − ∼10 10 mutations per base pair per generation, errors in MSH clamp at a mismatch can load multiple MLH clamps the nucleotide incorporation step occur about once every to license excision (1, 9). MutSα (MSH2-MSH6) and MutSβ + 104 to 105 insertions on average, although the frequency (MSH2-MSH3) are related to the AAA family of ATPases varies considerably depending on a number of parame- and carry out mismatch recognition (10, 11). MutL homo- ters including the particular mismatch, chromosomal con- logs MutLα (yeast ScMlh1-Pms1 and human HsMLH1- text, and the DNA polymerase. -

Exonuclease Activity and Removal of Bulky
Insight into mechanisms of 3′-5′ exonuclease activity PNAS PLUS and removal of bulky 8,5′-cyclopurine adducts by apurinic/apyrimidinic endonucleases Abdelghani Mazouzia, Armelle Vigourouxb, Bulat Aikesheva,c, Philip J. Brooksd,e, Murat K. Saparbaeva, Solange Morerab,1, and Alexander A. Ishchenkoa,1 aUniversité Paris-Sud, Laboratoire “Stabilité Génétique et Oncogenèse”, Centre National de la Recherche Scientifique (CNRS), Unité Mixte de Recherche 8200, Institut de Cancérologie Gustave-Roussy, F-94805 Villejuif Cedex, France; bLaboratoire d’Enzymologie et Biochimie Structurales, CNRS, F-91198 Gif-sur-Yvette Cedex, France; cL.N. Gumilev Eurasian National University, Astana, Republic of Kazakhstan, 010008; and dLaboratory of Neurogenetics and Division of Metabolism and Health Effects, National Institute on Alcohol Abuse and Alcoholism, eOffice of Rare Diseases Research, National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, MD 20892 Edited by Philip C. Hanawalt, Stanford University, Stanford, CA, and approved June 28, 2013 (received for review March 19, 2013) 8,5′-cyclo-2’-deoxyadenosine (cdA) and 8,5′-cyclo-2’-deoxyguano- compared with regular dA, implying that this base lesion would sine generated in DNA by both endogenous oxidative stress and be resistant to DNA glycosylase action (3). ionizing radiation are helix-distorting lesions and strong blocks for The cdA adducts in DNA are a strong block to various DNA DNA replication and transcription. In duplex DNA, these lesions are polymerases, such as T7, δ, and η (4). Interestingly, translesion repaired in the nucleotide excision repair (NER) pathway. How- DNA polymerase η can perform lesion bypass synthesis on the ever, lesions at DNA strand breaks are most likely poor substrates R-cdA but not on S-cdA (5). -

Exo1 Recruits Cdc5 Polo Kinase to Mutlγ to Ensure Efficient Meiotic Crossover Formation
Exo1 recruits Cdc5 polo kinase to MutLγ to ensure efficient meiotic crossover formation Aurore Sancheza,b,c, Céline Adama,b, Felix Rauhd, Yann Duroca,b, Lepakshi Ranjhac, Bérangère Lombarde, Xiaojing Muf,g, Mélody Wintreberta,b, Damarys Loewe, Alba Guarnéh, Stefano Gnana,b, Chun-Long Chena,b, Scott Keeneyf,g,i, Petr Cejkac, Raphaël Guéroisj, Franz Kleind, Jean-Baptiste Charbonnierj, and Valérie Bordea,b,1 aInstitut Curie, Paris Sciences et Lettres Research University, CNRS, UMR3244, 75005 Paris, France; bParis Sorbonne Université, UMR3244, 75005 Paris, France; cInstitute for Research in Biomedicine, Faculty of Biomedical Sciences, Università della Svizzera Italiana, 6501 Bellinzona, Switzerland; dMax F. Perutz Laboratories, University of Vienna, 1010 Wien, Austria; eInstitut Curie, Paris Sciences et Lettres Research University, 75006 Paris, France; fMolecular Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065; gWeill Graduate School of Medical Sciences, Cornell University, New York, NY 10065; hDepartment of Biochemistry, Centre de Recherche en Biologie Structurale, McGill University, Montreal, QC H3A 0G4, Canada; iMemorial Sloan Kettering Cancer Center, Howard Hughes Medical Institute, New York, NY 10065; and jInstitute for Integrative Biology of the Cell (I2BC), Commissariat à l’Energie Atomique, CNRS, Université Paris-Sud, Université Paris-Saclay, 91190 Gif-sur-Yvette, France Edited by Anne M. Villeneuve, Stanford University, Stanford, CA, and approved October 14, 2020 (received for review July 7, 2020) Crossovers generated during the repair of programmed meiotic recruits a MutL-related complex to initiate repair together with double-strand breaks must be tightly regulated to promote accu- proliferating cell nuclear antigen and Exo1 (reviewed in ref. 11). rate homolog segregation without deleterious outcomes, such as MutLα represents the major mismatch repair activity, which in- aneuploidy. -

Human Exonuclease 1 (EXO1) Regulatory Functions in DNA Replication with Putative Roles in Cancer
International Journal of Molecular Sciences Review Human Exonuclease 1 (EXO1) Regulatory Functions in DNA Replication with Putative Roles in Cancer Guido Keijzers 1,* , Daniela Bakula 1, Michael Angelo Petr 1,2, Nils Gedsig Kirkelund Madsen 1, Amanuel Teklu 1, Garik Mkrtchyan 1, Brenna Osborne 1 and Morten Scheibye-Knudsen 1 1 Department of Cellular and Molecular Medicine, Center for Healthy Aging, University of Copenhagen, DK-2200 Copenhagen, Denmark; [email protected] (D.B.); [email protected] (M.A.P.); [email protected] (N.G.K.M.); [email protected] (A.T.); [email protected] (G.M.); [email protected] (B.O.); [email protected] (M.S.-K.) 2 Experimental Gerontology Section, Translational Gerontology Branch, National Institute on Aging, NIH, Baltimore, MD 21224, USA * Correspondence: [email protected]; Tel.: +45-29-17-65-32 Received: 20 November 2018; Accepted: 19 December 2018; Published: 25 December 2018 Abstract: Human exonuclease 1 (EXO1), a 50!30 exonuclease, contributes to the regulation of the cell cycle checkpoints, replication fork maintenance, and post replicative DNA repair pathways. These processes are required for the resolution of stalled or blocked DNA replication that can lead to replication stress and potential collapse of the replication fork. Failure to restart the DNA replication process can result in double-strand breaks, cell-cycle arrest, cell death, or cellular transformation. In this review, we summarize the involvement of EXO1 in the replication, DNA repair pathways, cell cycle checkpoints, and the link between EXO1 and cancer. Keywords: DNA repair; double strand break repair; exonuclease 1; EXO1; mismatch repair; MMR; NER; nucleotide excision repair; strand displacements; TLS; translesion DNA synthesis 1.