The Human Exonuclease-1 Interactome and Phosphorylation Sites
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconstitution of Long and Short Patch Mismatch Repair Reactions Using Saccharomyces Cerevisiae Proteins
Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins Nikki Bowena, Catherine E. Smitha, Anjana Srivatsana, Smaranda Willcoxb,c, Jack D. Griffithb,c, and Richard D. Kolodnera,d,e,f,g,1 aLudwig Institute for Cancer Research, Departments of dMedicine and eCellular and Molecular Medicine, fMoores-University of California, San Diego Cancer Center, and gInstitute of Genomic Medicine, University of California San Diego School of Medicine, La Jolla, CA 92093; and bLineberger Cancer Center and cDepartment of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27514 Contributed by Richard D. Kolodner, October 8, 2013 (sent for review September 9, 2013) A problem in understanding eukaryotic DNA mismatch repair In eukaryotic MMR, mispairs are bound by MutS homolog 2 (MMR) mechanisms is linking insights into MMR mechanisms from (Msh2)–MutS homolog 6 (Msh6) and Msh2–MutS homolog 3 genetics and cell-biology studies with those from biochemical (Msh3), two partially redundant complexes of MutS-related pro- studies of MMR proteins and reconstituted MMR reactions. This teins (3, 4, 18, 19). These complexes recruit a MutL-related type of analysis has proven difficult because reconstitution ap- complex, called MutL homoloh 1 (Mlh1)–postmeiotic segrega- proaches have been most successful for human MMR whereas tion 1 (Pms1) in S. cerevisiae and Mlh1–postmeiotic segregation – – analysis of MMR in vivo has been most advanced in the yeast 2 (Pms2) in human and mouse (3, 4, 20 23). The Mlh1 Pms1/ Saccharomyces cerevisiae. Here, we describe the reconstitution of Pms2 complex has an endonuclease activity suggested to play MMR reactions using purified S. -

Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA
Mechanisms in E. coli and Human Mismatch Repair Nobel Lecture, December 8, 2015 by Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA. he idea that mismatched base pairs occur in cells and that such lesions trig- T ger their own repair was suggested 50 years ago by Robin Holliday in the context of genetic recombination [1]. Breakage and rejoining of DNA helices was known to occur during this process [2], with precision of rejoining attributed to formation of a heteroduplex joint, a region of helix where the two strands are derived from the diferent recombining partners. Holliday pointed out that if this heteroduplex region should span a genetic diference between the two DNAs, then it will contain one or more mismatched base pairs. He invoked processing of such mismatches to explain the recombination-associated phenomenon of gene conversion [1], noting that “If there are enzymes which can repair points of damage in DNA, it would seem possible that the same enzymes could recognize the abnormality of base pairing, and by exchange reactions rectify this.” Direct evidence that mismatches provoke a repair reaction was provided by bacterial transformation experiments [3–5], and our interest in this efect was prompted by the Escherichia coli (E. coli) work done in Matt Meselson’s lab at Harvard. Using artifcially constructed heteroduplex DNAs containing multiple mismatched base pairs, Wagner and Meselson [6] demonstrated that mismatches elicit a repair reaction upon introduction into the E. coli cell. Tey also showed that closely spaced mismatches, mismatches separated by a 1000 base pairs or so, are usually repaired on the same DNA strand. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Restriction Endonucleases
Molecular Biology Problem Solver: A Laboratory Guide. Edited by Alan S. Gerstein Copyright © 2001 by Wiley-Liss, Inc. ISBNs: 0-471-37972-7 (Paper); 0-471-22390-5 (Electronic) 9 Restriction Endonucleases Derek Robinson, Paul R. Walsh, and Joseph A. Bonventre Background Information . 226 Which Restriction Enzymes Are Commercially Available? . 226 Why Are Some Enzymes More Expensive Than Others? . 227 What Can You Do to Reduce the Cost of Working with Restriction Enzymes? . 228 If You Could Select among Several Restriction Enzymes for Your Application, What Criteria Should You Consider to Make the Most Appropriate Choice? . 229 What Are the General Properties of Restriction Endonucleases? . 232 What Insight Is Provided by a Restriction Enzyme’s Quality Control Data? . 233 How Stable Are Restriction Enzymes? . 236 How Stable Are Diluted Restriction Enzymes? . 236 Simple Digests . 236 How Should You Set up a Simple Restriction Digest? . 236 Is It Wise to Modify the Suggested Reaction Conditions? . 237 Complex Restriction Digestions . 239 How Can a Substrate Affect the Restriction Digest? . 239 Should You Alter the Reaction Volume and DNA Concentration? . 241 Double Digests: Simultaneous or Sequential? . 242 225 Genomic Digests . 244 When Preparing Genomic DNA for Southern Blotting, How Can You Determine If Complete Digestion Has Been Obtained? . 244 What Are Your Options If You Must Create Additional Rare or Unique Restriction Sites? . 247 Troubleshooting . 255 What Can Cause a Simple Restriction Digest to Fail? . 255 The Volume of Enzyme in the Vial Appears Very Low. Did Leakage Occur during Shipment? . 259 The Enzyme Shipment Sat on the Shipping Dock for Two Days. -

Phosphate Steering by Flap Endonuclease 1 Promotes 50-flap Specificity and Incision to Prevent Genome Instability
ARTICLE Received 18 Jan 2017 | Accepted 5 May 2017 | Published 27 Jun 2017 DOI: 10.1038/ncomms15855 OPEN Phosphate steering by Flap Endonuclease 1 promotes 50-flap specificity and incision to prevent genome instability Susan E. Tsutakawa1,*, Mark J. Thompson2,*, Andrew S. Arvai3,*, Alexander J. Neil4,*, Steven J. Shaw2, Sana I. Algasaier2, Jane C. Kim4, L. David Finger2, Emma Jardine2, Victoria J.B. Gotham2, Altaf H. Sarker5, Mai Z. Her1, Fahad Rashid6, Samir M. Hamdan6, Sergei M. Mirkin4, Jane A. Grasby2 & John A. Tainer1,7 DNA replication and repair enzyme Flap Endonuclease 1 (FEN1) is vital for genome integrity, and FEN1 mutations arise in multiple cancers. FEN1 precisely cleaves single-stranded (ss) 50-flaps one nucleotide into duplex (ds) DNA. Yet, how FEN1 selects for but does not incise the ss 50-flap was enigmatic. Here we combine crystallographic, biochemical and genetic analyses to show that two dsDNA binding sites set the 50polarity and to reveal unexpected control of the DNA phosphodiester backbone by electrostatic interactions. Via ‘phosphate steering’, basic residues energetically steer an inverted ss 50-flap through a gateway over FEN1’s active site and shift dsDNA for catalysis. Mutations of these residues cause an 18,000-fold reduction in catalytic rate in vitro and large-scale trinucleotide (GAA)n repeat expansions in vivo, implying failed phosphate-steering promotes an unanticipated lagging-strand template-switch mechanism during replication. Thus, phosphate steering is an unappreciated FEN1 function that enforces 50-flap specificity and catalysis, preventing genomic instability. 1 Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Structure and Function of Nucleases in DNA Repair: Shape, Grip and Blade of the DNA Scissors
Oncogene (2002) 21, 9022 – 9032 ª 2002 Nature Publishing Group All rights reserved 0950 – 9232/02 $25.00 www.nature.com/onc Structure and function of nucleases in DNA repair: shape, grip and blade of the DNA scissors Tatsuya Nishino1 and Kosuke Morikawa*,1 1Department of Structural Biology, Biomolecular Engineering Research Institute (BERI), 6-2-3 Furuedai, Suita, Osaka 565-0874, Japan DNA nucleases catalyze the cleavage of phosphodiester mismatched nucleotides. They also recognize the bonds. These enzymes play crucial roles in various DNA replication or recombination intermediates to facilitate repair processes, which involve DNA replication, base the following reaction steps through the cleavage of excision repair, nucleotide excision repair, mismatch DNA strands (Table 1). repair, and double strand break repair. In recent years, Nucleases can be regarded as molecular scissors, new nucleases involved in various DNA repair processes which cleave phosphodiester bonds between the sugars have been reported, including the Mus81 : Mms4 (Eme1) and the phosphate moieties of DNA. They contain complex, which functions during the meiotic phase and conserved minimal motifs, which usually consist of the Artemis : DNA-PK complex, which processes a V(D)J acidic and basic residues forming the active site. recombination intermediate. Defects of these nucleases These active site residues coordinate catalytically cause genetic instability or severe immunodeficiency. essential divalent cations, such as magnesium, Thus, structural biology on various nuclease actions is calcium, manganese or zinc, as a cofactor. However, essential for the elucidation of the molecular mechanism the requirements for actual cleavage, such as the types of complex DNA repair machinery. Three-dimensional and the numbers of metals, are very complicated, but structural information of nucleases is also rapidly are not common among the nucleases. -

Flap DNA Unwinding and Incision by the Human FAN1 Dimer
ARTICLE Received 16 Aug 2014 | Accepted 30 Oct 2014 | Published 11 Dec 2014 DOI: 10.1038/ncomms6726 Structural insights into 50 flap DNA unwinding and incision by the human FAN1 dimer Qi Zhao1,*, Xiaoyu Xue1,*, Simonne Longerich1,*, Patrick Sung1 & Yong Xiong1 Human FANCD2-associated nuclease 1 (FAN1) is a DNA structure-specific nuclease involved in the processing of DNA interstrand crosslinks (ICLs). FAN1 maintains genomic stability and prevents tissue decline in multiple organs, yet it confers ICL-induced anti-cancer drug resistance in several cancer subtypes. Here we report three crystal structures of human FAN1 in complex with a 50 flap DNA substrate, showing that two FAN1 molecules form a head-to- tail dimer to locate the lesion, orient the DNA and unwind a 50 flap for subsequent incision. Biochemical experiments further validate our model for FAN1 action, as structure-informed mutations that disrupt protein dimerization, substrate orientation or flap unwinding impair the structure-specific nuclease activity. Our work elucidates essential aspects of FAN1-DNA lesion recognition and a unique mechanism of incision. These structural insights shed light on the cellular mechanisms underlying organ degeneration protection and cancer drug resistance mediated by FAN1. 1 Department of Molecular Biophysics and Biochemistry, Yale University School of Medicine, New Haven, Connecticut 06520, USA. * These authors contributed equally to this work. Correspondence and requests for materials should be addressed to P.S. (email: [email protected]) or to Y.X. (email: [email protected]). NATURE COMMUNICATIONS | 5:5726 | DOI: 10.1038/ncomms6726 | www.nature.com/naturecommunications 1 & 2014 Macmillan Publishers Limited. All rights reserved. -

Datasheet for Exonuclease V (Recbcd)
Source: An E. coli strain containing plasmids Unit Assay Conditions: 1X NEBuffer 4, 1 mM ATP Physical Purity: Purified to > 95% homogene- Exonuclease V for expressing the three subunits of E. coli with 0.15 mM sonicated duplex [3H]-DNA. ity as determined by SDS-PAGE analysis using Exonuclease V: RecB, RecC and RecD. Coomassie Blue detection. (RecBCD) Heat Inactivation: 70°C for 30 minutes. Supplied in: 100 mM NaCl, 50 mM Tris-HCl A Typical Exonuclease V Reaction: 1-800-632-7799 (pH 7.5 @ 25°C), 0.1 mM EDTA, 1 mM DTT, Quality Control Assays x µl sample DNA (~ 1 µg) [email protected] 0.1% Triton X-100 and 50% glycerol. 3 µl NEBuffer4 (10X) www.neb.com Endonuclease Activity I: Incubation of a 50 µl 3 µl 10 mM ATP M0345S 001121014101 reaction containing 100 units of Exonuclease V Reagents Supplied with Enzyme: y µl H20 (up to final volume of 30 µl) 10X NEBuffer 4, 10 mM ATP with 1 µg of φX174 RF I DNA in NEBuffer 4 and 1 µl Exonuclease V (10 units) 1 mM ATP for 4 hours at 37°C resulted in < 10% M0345S Reaction Conditions: 1X NEBuffer 4 loss in φX174 RF I DNA as determined by agarose 1. Incubate at 37°C for 30 minutes. 2. To stop reaction add EDTA to 11 mM. 1,000 units 10,000 U/ml Lot: 0011210 supplemented with 1 mM ATP. Incubate at 37°C. gel electrophoresis. 3 Heat Inactivation 70°C for 30 minutes. RECOMBINANT Store at –20°C Exp: 10/14 1X NEBuffer 4: Endonuclease Activity II: Incubation of a 50 µl 4. -

Complete Genome of the Cellulolytic Thermophile Acidothermus Cellulolyticus 11B Provides Insights Into Its Ecophysiological and Evolutionary Adaptations
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations Ravi D. Barabote,1,9 Gary Xie,1 David H. Leu,2 Philippe Normand,3 Anamaria Necsulea,4 Vincent Daubin,4 Claudine Me´digue,5 William S. Adney,6 Xin Clare Xu,2 Alla Lapidus,7 Rebecca E. Parales,8 Chris Detter,1 Petar Pujic,3 David Bruce,1 Celine Lavire,3 Jean F. Challacombe,1 Thomas S. Brettin,1 and Alison M. Berry2,10 1DOE Joint Genome Institute, Bioscience Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA; 2Department of Plant Sciences, University of California, Davis, California 95616, USA; 3Centre National de la Recherche Scientifique (CNRS), UMR5557, E´cologie Microbienne, Universite´ Lyon I, Villeurbanne F-69622, France; 4Centre National de la Recherche Scientifique (CNRS), UMR5558, Laboratoire de Biome´trie et Biologie E´volutive, Universite´ Lyon I, Villeurbanne F-69622, France; 5Centre National de la Recherche Scientifique (CNRS), UMR8030 and CEA/DSV/IG/Genoscope, Laboratoire de Ge´nomique Comparative, 91057 Evry Cedex, France; 6National Renewable Energy Laboratory, Golden, Colorado 80401, USA; 7DOE Joint Genome Institute, Walnut Creek, California 94598, USA; 8Department of Microbiology, University of California, Davis, California 95616, USA We present here the complete 2.4-Mb genome of the cellulolytic actinobacterial thermophile Acidothermus cellulolyticus 11B. New secreted glycoside hydrolases and carbohydrate esterases were identified in the genome, revealing a diverse biomass- degrading enzyme repertoire far greater than previously characterized and elevating the industrial value of this organism. -
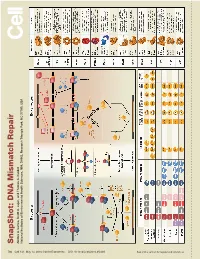
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Using Exonuclease V (Recbcd) to Eliminate Residual Genomic DNA When Purifying Low Copy Plasmids with the Monarch® Plasmid Miniprep Kit
be INSPIRED APPLICATION NOTE drive DISCOVERY stay GENUINE Using Exonuclease V (RecBCD) to Eliminate Residual Genomic DNA When Purifying Low Copy Plasmids with the Monarch® Plasmid Miniprep Kit Peichung Hsieh, Ph.D., New England Biolabs, Inc. Introduction Protocol Materials The use of low and/or single-copy plasmids 1. Transform an endA- strain (e.g. NEB 10-beta, Endonuclease V (RecBCD) (NEB #M0345) to clone large pieces of DNA (up to 200 kb) NEB #C3019) with the BAC plasmid DNA or to drive expression of slow folding or toxic and plate outgrowth onto a media plate with NEB 10-beta Competent E coli (High Efficiency)(NEB #C3019) proteins in E.coli is a commonly used strategy. appropriate antibiotic. Incubate overnight Antibiotic, typically Chloramphenicol Purification of low-copy plasmids or bacterial at 30°C. BACs with CamR require reduced artificial chromosomes (BACs) presents some stringency selection. Chloramphenicol levels LB Media challenges that are not evident when working should be maintained between Monarch Plasmid Miniprep Kit (NEB #T1010) with higher copy number plasmids such as 10-15 μg/ml on the selective plate. pUC19. The ratio between bacterial gDNA Note: strains with an F’ plasmid are not compatible and plasmid DNA is higher, thereby reducing with BACs or miniF plasmids. yield of the desired plasmid produced 2. Pick a colony, inoculate 10 ml LB + by typical plasmid miniprep protocols. antibiotic, and incubate overnight at 30°C Additionally, elevated levels of host gDNA (200-250 RPM). are often co-purified, reducing the accuracy 3. Check OD600 nm (usually it will be around 4 of quantitation by UV absorbance or dsDNA O.D./ml of cells).