3 Exoribonucleases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconstitution of Long and Short Patch Mismatch Repair Reactions Using Saccharomyces Cerevisiae Proteins
Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins Nikki Bowena, Catherine E. Smitha, Anjana Srivatsana, Smaranda Willcoxb,c, Jack D. Griffithb,c, and Richard D. Kolodnera,d,e,f,g,1 aLudwig Institute for Cancer Research, Departments of dMedicine and eCellular and Molecular Medicine, fMoores-University of California, San Diego Cancer Center, and gInstitute of Genomic Medicine, University of California San Diego School of Medicine, La Jolla, CA 92093; and bLineberger Cancer Center and cDepartment of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27514 Contributed by Richard D. Kolodner, October 8, 2013 (sent for review September 9, 2013) A problem in understanding eukaryotic DNA mismatch repair In eukaryotic MMR, mispairs are bound by MutS homolog 2 (MMR) mechanisms is linking insights into MMR mechanisms from (Msh2)–MutS homolog 6 (Msh6) and Msh2–MutS homolog 3 genetics and cell-biology studies with those from biochemical (Msh3), two partially redundant complexes of MutS-related pro- studies of MMR proteins and reconstituted MMR reactions. This teins (3, 4, 18, 19). These complexes recruit a MutL-related type of analysis has proven difficult because reconstitution ap- complex, called MutL homoloh 1 (Mlh1)–postmeiotic segrega- proaches have been most successful for human MMR whereas tion 1 (Pms1) in S. cerevisiae and Mlh1–postmeiotic segregation – – analysis of MMR in vivo has been most advanced in the yeast 2 (Pms2) in human and mouse (3, 4, 20 23). The Mlh1 Pms1/ Saccharomyces cerevisiae. Here, we describe the reconstitution of Pms2 complex has an endonuclease activity suggested to play MMR reactions using purified S. -

Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA
Mechanisms in E. coli and Human Mismatch Repair Nobel Lecture, December 8, 2015 by Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA. he idea that mismatched base pairs occur in cells and that such lesions trig- T ger their own repair was suggested 50 years ago by Robin Holliday in the context of genetic recombination [1]. Breakage and rejoining of DNA helices was known to occur during this process [2], with precision of rejoining attributed to formation of a heteroduplex joint, a region of helix where the two strands are derived from the diferent recombining partners. Holliday pointed out that if this heteroduplex region should span a genetic diference between the two DNAs, then it will contain one or more mismatched base pairs. He invoked processing of such mismatches to explain the recombination-associated phenomenon of gene conversion [1], noting that “If there are enzymes which can repair points of damage in DNA, it would seem possible that the same enzymes could recognize the abnormality of base pairing, and by exchange reactions rectify this.” Direct evidence that mismatches provoke a repair reaction was provided by bacterial transformation experiments [3–5], and our interest in this efect was prompted by the Escherichia coli (E. coli) work done in Matt Meselson’s lab at Harvard. Using artifcially constructed heteroduplex DNAs containing multiple mismatched base pairs, Wagner and Meselson [6] demonstrated that mismatches elicit a repair reaction upon introduction into the E. coli cell. Tey also showed that closely spaced mismatches, mismatches separated by a 1000 base pairs or so, are usually repaired on the same DNA strand. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Phosphate Steering by Flap Endonuclease 1 Promotes 50-flap Specificity and Incision to Prevent Genome Instability
ARTICLE Received 18 Jan 2017 | Accepted 5 May 2017 | Published 27 Jun 2017 DOI: 10.1038/ncomms15855 OPEN Phosphate steering by Flap Endonuclease 1 promotes 50-flap specificity and incision to prevent genome instability Susan E. Tsutakawa1,*, Mark J. Thompson2,*, Andrew S. Arvai3,*, Alexander J. Neil4,*, Steven J. Shaw2, Sana I. Algasaier2, Jane C. Kim4, L. David Finger2, Emma Jardine2, Victoria J.B. Gotham2, Altaf H. Sarker5, Mai Z. Her1, Fahad Rashid6, Samir M. Hamdan6, Sergei M. Mirkin4, Jane A. Grasby2 & John A. Tainer1,7 DNA replication and repair enzyme Flap Endonuclease 1 (FEN1) is vital for genome integrity, and FEN1 mutations arise in multiple cancers. FEN1 precisely cleaves single-stranded (ss) 50-flaps one nucleotide into duplex (ds) DNA. Yet, how FEN1 selects for but does not incise the ss 50-flap was enigmatic. Here we combine crystallographic, biochemical and genetic analyses to show that two dsDNA binding sites set the 50polarity and to reveal unexpected control of the DNA phosphodiester backbone by electrostatic interactions. Via ‘phosphate steering’, basic residues energetically steer an inverted ss 50-flap through a gateway over FEN1’s active site and shift dsDNA for catalysis. Mutations of these residues cause an 18,000-fold reduction in catalytic rate in vitro and large-scale trinucleotide (GAA)n repeat expansions in vivo, implying failed phosphate-steering promotes an unanticipated lagging-strand template-switch mechanism during replication. Thus, phosphate steering is an unappreciated FEN1 function that enforces 50-flap specificity and catalysis, preventing genomic instability. 1 Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -

The Roles of the Exoribonucleases DIS3L2 and XRN1 in Human Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Sussex Research Online The roles of the exoribonucleases DIS3L2 and XRN1 in human disease. Amy L. Pashler1, Ben Towler1, Christopher I. Jones2 and Sarah F. Newbury* 1Brighton and Sussex Medical School, Medical Research building, University of Sussex, Falmer, Brighton, BN1 9PS, U.K. 2Brighton and Sussex Medical School, Department of Primary Care and Public Health, University of Brighton, Falmer, Brighton, BN1 9PH, U.K. *Corresponding author: [email protected] +44 (0)1273 877874 Abstract RNA degradation is a vital post-transcriptional process which ensures that transcripts are maintained at the correct level within the cell. DIS3L2 and XRN1 are conserved exoribonucleases which are critical for the degradation of cytoplasmic RNAs. Although the molecular mechanisms of RNA degradation by DIS3L2 and XRN1 have been well studied, less is known about their specific roles in development of multicellular organisms or human disease. This review focusses on the roles of DIS3L2 and XRN1 in the pathogenesis of human disease, particularly in relation to phenotypes seen in model organisms. The known diseases associated with loss of activity of DIS3L2 and XRN1 are discussed, together with possible mechanisms and cellular pathways leading to these disease conditions. Key words: RNA stability, RNA degradation, human disease, virus-host interactions, XRN1, DIS3L2 Abbreviations used: UTR, untranslated region; HCV, Hepatitis C virus; sfRNA, small flaviviral non- coding RNA; mRNA, messenger RNA; miRNA, microRNA. Introduction Ribonucleases are key enzymes involved in the control of mRNA stability, which is crucially important in maintaining RNA transcripts at the correct levels within cells. -

Xrn1 - a Major Mrna Synthesis and Decay Factor
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.01.437949; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. RNA-controlled nucleocytoplasmic shuttling of mRNA decay factors regulates mRNA synthesis and initiates a novel mRNA decay pathway Running title: Cytoplasmic mRNA decay begins in the nucleus Shiladitya Chattopadhyay1, Jose Garcia-Martinez2, Gal Haimovich1,3, Aya Khwaja1, Oren Barkai1, Ambarnil Ghosh4, Silvia Gabriela Chuarzman5, Maya Schuldiner5, Ron Elran1, Miriam Rosenberg1, Katherine Bohnsack6, Markus Bohnsack6, Jose E Perez-Ortin2, Mordechai Choder1* 1Department of Molecular Microbiology, Rappaport Faculty of Medicine, Technion-Israel Institute of Technology, Haifa 31096, Israel. 2Instituto de Biotecnología y Biomedicina (Biotecmed), Universitat de València ; Burjassot, Valencia, 46100, Spain. 3current address:Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 7610001, Israel. 4UCD Conway Institute of Biomolecular & Biomedical Research, Dublin, Ireland. 5Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 7610001, Israel 6Institute for Molecular Biology, University Medical Center Göttingen Georg-August- University, Göttingen, Germany Göttingen Centre for Molecular Biosciences, Georg-August- University, Göttingen, Germany *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.01.437949; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Effect of Cigarette Smoke on DNA Methylation and Lung Function
de Vries et al. Respiratory Research (2018) 19:212 https://doi.org/10.1186/s12931-018-0904-y RESEARCH Open Access From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function Maaike de Vries1,2* , Diana A van der Plaat1,2, Ivana Nedeljkovic3, Rikst Nynke Verkaik-Schakel4, Wierd Kooistra2,5, Najaf Amin3, Cornelia M van Duijn3, Corry-Anke Brandsma2,5, Cleo C van Diemen6, Judith M Vonk1,2 and H Marike Boezen1,2 Abstract Background: Genetic and environmental factors play a role in the development of COPD. The epigenome, and more specifically DNA methylation, is recognized as important link between these factors. We postulate that DNA methylation is one of the routes by which cigarette smoke influences the development of COPD. In this study, we aim to identify CpG-sites that are associated with cigarette smoke exposure and lung function levels in whole blood and validate these CpG-sites in lung tissue. Methods: The association between pack years and DNA methylation was studied genome-wide in 658 current smokers with >5 pack years using robust linear regression analysis. Using mediation analysis, we subsequently selected the CpG-sites that were also associated with lung function levels. Significant CpG-sites were validated in lung tissue with pyrosequencing and expression quantitative trait methylation (eQTM) analysis was performed to investigate the association between DNA methylation and gene expression. Results: 15 CpG-sites were significantly associated with pack years and 10 of these were additionally associated with lung function levels. We validated 5 CpG-sites in lung tissue and found several associations between DNA methylation and gene expression. -
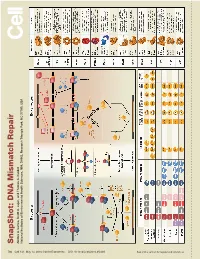
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Genome-Wide Analyses of XRN1-Sensitive Targets in Osteosarcoma Cells Identifies Disease-Relevant 2 Transcripts Containing G-Rich Motifs
Downloaded from rnajournal.cshlp.org on October 11, 2021 - Published by Cold Spring Harbor Laboratory Press 1 Genome-wide analyses of XRN1-sensitive targets in osteosarcoma cells identifies disease-relevant 2 transcripts containing G-rich motifs. 3 Amy L. Pashler, Benjamin P. Towler+, Christopher I. Jones, Hope J. Haime, Tom Burgess, and Sarah F. 4 Newbury1+ 5 6 Brighton and Sussex Medical School, University of Sussex, Brighton, BN1 9PS, UK 7 8 +Corresponding author: Prof Sarah Newbury, Medical Research Building, Brighton and Sussex 9 Medical School, University of Sussex, Falmer, Brighton BN1 9PS, UK. 10 Tel: +44(0)1273 877874 11 [email protected] 12 13 +Co-corresponding author: Dr Ben Towler, Medical Research Building, Brighton and Sussex Medical 14 School, University of Sussex, Falmer, Brighton BN1 9PS, UK. 15 Tel: +44(0)1273 877876 16 [email protected] 17 18 Running title: Genome-wide analyses of XRN1 targets in OS cells 19 20 Key words: XRN1, RNA-seq, Ewing sarcoma, lncRNAs, RNA degradation 21 22 23 24 25 26 27 28 29 30 31 32 33 Pashler et al 1 Downloaded from rnajournal.cshlp.org on October 11, 2021 - Published by Cold Spring Harbor Laboratory Press 34 35 ABSTRACT 36 XRN1 is a highly conserved exoribonuclease which degrades uncapped RNAs in a 5’-3’ direction. 37 Degradation of RNAs by XRN1 is important in many cellular and developmental processes and is 38 relevant to human disease. Studies in D. melanogaster demonstrate that XRN1 can target specific 39 RNAs, which have important consequences for developmental pathways. -

Differential Expression Profile Prioritization of Positional Candidate Glaucoma Genes the GLC1C Locus
LABORATORY SCIENCES Differential Expression Profile Prioritization of Positional Candidate Glaucoma Genes The GLC1C Locus Frank W. Rozsa, PhD; Kathleen M. Scott, BS; Hemant Pawar, PhD; John R. Samples, MD; Mary K. Wirtz, PhD; Julia E. Richards, PhD Objectives: To develop and apply a model for priori- est because of moderate expression and changes in tization of candidate glaucoma genes. expression. Transcription factor ZBTB38 emerges as an interesting candidate gene because of the overall expres- Methods: This Affymetrix GeneChip (Affymetrix, Santa sion level, differential expression, and function. Clara, Calif) study of gene expression in primary cul- ture human trabecular meshwork cells uses a positional Conclusions: Only1geneintheGLC1C interval fits our differential expression profile model for prioritization of model for differential expression under multiple glau- candidate genes within the GLC1C genetic inclusion in- coma risk conditions. The use of multiple prioritization terval. models resulted in filtering 7 candidate genes of higher interest out of the 41 known genes in the region. Results: Sixteen genes were expressed under all condi- tions within the GLC1C interval. TMEM22 was the only Clinical Relevance: This study identified a small sub- gene within the interval with differential expression in set of genes that are most likely to harbor mutations that the same direction under both conditions tested. Two cause glaucoma linked to GLC1C. genes, ATP1B3 and COPB2, are of interest in the con- text of a protein-misfolding model for candidate selec- tion. SLC25A36, PCCB, and FNDC6 are of lesser inter- Arch Ophthalmol. 2007;125:117-127 IGH PREVALENCE AND PO- identification of additional GLC1C fami- tential for severe out- lies7,18-20 who provide optimal samples for come combine to make screening candidate genes for muta- adult-onset primary tions.7,18,20 The existence of 2 distinct open-angle glaucoma GLC1C haplotypes suggests that muta- (POAG) a significant public health prob- tions will not be limited to rare descen- H1 lem.