Projet De Nouvelle Page Titre
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Relevance of Clinical, Genetic and Serological Markers
AUTREV-01901; No of Pages 18 Autoimmunity Reviews xxx (2016) xxx–xxx Contents lists available at ScienceDirect Autoimmunity Reviews journal homepage: www.elsevier.com/locate/autrev Review Cardiovascular risk assessment in patients with rheumatoid arthritis: The relevance of clinical, genetic and serological markers Raquel López-Mejías a, Santos Castañeda b, Carlos González-Juanatey c,AlfonsoCorralesa, Iván Ferraz-Amaro d, Fernanda Genre a, Sara Remuzgo-Martínez a, Luis Rodriguez-Rodriguez e, Ricardo Blanco a,JavierLlorcaf, Javier Martín g, Miguel A. González-Gay a,h,i,⁎ a Epidemiology, Genetics and Atherosclerosis Research Group on Systemic Inflammatory Diseases, Rheumatology Division, IDIVAL, Santander, Spain b Division of Rheumatology, Hospital Universitario la Princesa, IIS-IPrincesa, Madrid, Spain c Division of Cardiology, Hospital Lucus Augusti, Lugo, Spain d Rheumatology Division, Hospital Universitario de Canarias, Santa Cruz de Tenerife, Spain e Division of Rheumatology, Hospital Clínico San Carlos, Madrid, Spain f Division of Epidemiology and Computational Biology, School of Medicine, University of Cantabria, and CIBER Epidemiología y Salud Pública (CIBERESP), IDIVAL, Santander, Spain g Institute of Parasitology and Biomedicine López-Neyra, IPBLN-CSIC, Granada, Spain h School of Medicine, University of Cantabria, Santander, Spain i Cardiovascular Pathophysiology and Genomics Research Unit, School of Physiology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa article info abstract Article history: Cardiovascular disease (CV) is the most common cause of premature mortality in patients with rheumatoid ar- Received 7 July 2016 thritis (RA). This is the result of an accelerated atherosclerotic process. Adequate CV risk stratification has special Accepted 9 July 2016 relevance in RA to identify patients at risk of CV disease. -

Supp Material.Pdf
Simon et al. Supplementary information: Table of contents p.1 Supplementary material and methods p.2-4 • PoIy(I)-poly(C) Treatment • Flow Cytometry and Immunohistochemistry • Western Blotting • Quantitative RT-PCR • Fluorescence In Situ Hybridization • RNA-Seq • Exome capture • Sequencing Supplementary Figures and Tables Suppl. items Description pages Figure 1 Inactivation of Ezh2 affects normal thymocyte development 5 Figure 2 Ezh2 mouse leukemias express cell surface T cell receptor 6 Figure 3 Expression of EZH2 and Hox genes in T-ALL 7 Figure 4 Additional mutation et deletion of chromatin modifiers in T-ALL 8 Figure 5 PRC2 expression and activity in human lymphoproliferative disease 9 Figure 6 PRC2 regulatory network (String analysis) 10 Table 1 Primers and probes for detection of PRC2 genes 11 Table 2 Patient and T-ALL characteristics 12 Table 3 Statistics of RNA and DNA sequencing 13 Table 4 Mutations found in human T-ALLs (see Fig. 3D and Suppl. Fig. 4) 14 Table 5 SNP populations in analyzed human T-ALL samples 15 Table 6 List of altered genes in T-ALL for DAVID analysis 20 Table 7 List of David functional clusters 31 Table 8 List of acquired SNP tested in normal non leukemic DNA 32 1 Simon et al. Supplementary Material and Methods PoIy(I)-poly(C) Treatment. pIpC (GE Healthcare Lifesciences) was dissolved in endotoxin-free D-PBS (Gibco) at a concentration of 2 mg/ml. Mice received four consecutive injections of 150 μg pIpC every other day. The day of the last pIpC injection was designated as day 0 of experiment. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

Original Article Association Study of BUD13-ZNF259 Gene Rs964184 Polymorphism and Hemorrhagic Stroke Risk
Int J Clin Exp Med 2015;8(12):22503-22508 www.ijcem.com /ISSN:1940-5901/IJCEM0016028 Original Article Association study of BUD13-ZNF259 gene rs964184 polymorphism and hemorrhagic stroke risk Shengjun Zhou, Jikuang Zhao, Zhepei Wang, Keqin Li, Sheng Nie, Feng Gao, Jie Sun, Xiang Gao, Yi Huang Department of Neurosurgery, Ningbo First Hospital, Ningbo Hospital of Zhejiang University, Ningbo 315010, Zhe- jiang, China Received September 11, 2015; Accepted December 3, 2015; Epub December 15, 2015; Published December 30, 2015 Abstract: We aimed to evaluate the association of rs964184 of BUD13-ZNF259 gene with the risk of hemorrhagic stroke (HS). A total of 138 HS cases and 587 controls were recruited for the association of rs964184 of BUD13- ZNF259 gene with the risk of HS. Tm shift PCR was used for genotyping. We were unable to find the association of rs964184 of BUD13-ZNF259 gene with the risk of HS (P>0.05). Significant difference was found in the TG level among the three genotypes (CC: 1.51±1.02; CG: 1.68±1.10; GG: 1.90±1.11, P=0.036). The TG level showed strong correlation with rs964184 genotypes (P=0.010, correlation=0.101). Significantly higher TC, HDL-C, and LDL-C levels were observed in the case group. And no difference was found in the TG, ApoA-I, ApoB. Our case-control study sup- ported the significant association between rs964184 genotype and the blood TG concentration, although we were unable to find association between BUD13-ZNF259 rs964184 and the risk of HS in Han Chinese. -

ZNF259 Inhibits Non-Small Cell Lung Cancer Cells Proliferation and Invasion by FAK-AKT Signaling
Journal name: Cancer Management and Research Article Designation: ORIGINAL RESEARCH Year: 2017 Volume: 9 Cancer Management and Research Dovepress Running head verso: Shan et al Running head recto: ZNF259 inhibits NSCLC proliferation and invasion open access to scientific and medical research DOI: http://dx.doi.org/10.2147/CMAR.S150614 Open Access Full Text Article ORIGINAL RESEARCH ZNF259 inhibits non-small cell lung cancer cells proliferation and invasion by FAK-AKT signaling Yuemei Shan1,2 Background: Zinc finger protein 259 (ZNF259) is known to play essential roles in embryonic Wei Cao1 development and cell cycle regulation. However, its expression pattern and clinicopathological Tao Wang1 relevance remain unclear. Guiyang Jiang3 Materials and methods: A total of 114 lung cancer specimens were collected. The ZNF259 Yong Zhang4 expression was measured between the lung cancer tissues and the adjacent normal lung tissues by immunohistochemical staining and Western blotting. Moreover, the correlation of ZNF259 Xianghong Yang1 expression with clinicopathological features was analyzed in 114 cases of lung cancer. Addition- 1 Department of Pathology, ally, ZNF259 was depleted in the lung cancer cells in order to analyze its effect in the lung cancer. Shengjing Hospital of China Medical University, Shenyang, China; Results: Immunohistochemical staining of 114 lung cancer specimens revealed significantly 2 For personal use only. Department of Applied Technology, lower ZNF259 expression in lung cancer tissues than in adjacent normal lung tissues (53.5% vs Institute of Technology of China 71.4%, P<0.001). In addition, ZNF259 downregulation was significantly associated with larger Medical University, Shenyang, China; 3Department of Pathology, First tumor size (P=0.001), advanced TNM stage (P=0.002), and positive lymph node metastasis Affiliated Hospital and College (P=0.02). -

Download Special Issue
BioMed Research International Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data Guest Editors: Julia Tzu-Ya Weng, Li-Ching Wu, Wen-Chi Chang, Tzu-Hao Chang, Tatsuya Akutsu, and Tzong-Yi Lee Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data BioMed Research International Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data Guest Editors: Julia Tzu-Ya Weng, Li-Ching Wu, Wen-Chi Chang, Tzu-Hao Chang, Tatsuya Akutsu, and Tzong-Yi Lee Copyright © 2014 Hindawi Publishing Corporation. All rights reserved. This is a special issue published in “BioMed Research International.” All articles are open access articles distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Contents Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data,JuliaTzu-YaWeng, Li-Ching Wu, Wen-Chi Chang, Tzu-Hao Chang, Tatsuya Akutsu, and Tzong-Yi Lee Volume2014,ArticleID814092,3pages Evolution of Network Biomarkers from Early to Late Stage Bladder Cancer Samples,Yung-HaoWong, Cheng-Wei Li, and Bor-Sen Chen Volume 2014, Article ID 159078, 23 pages MicroRNA Expression Profiling Altered by Variant Dosage of Radiation Exposure,Kuei-FangLee, Yi-Cheng Chen, Paul Wei-Che Hsu, Ingrid Y. Liu, and Lawrence Shih-Hsin Wu Volume2014,ArticleID456323,10pages EXIA2: Web Server of Accurate and Rapid Protein Catalytic Residue Prediction, Chih-Hao Lu, Chin-Sheng -

The Kinesin Spindle Protein Inhibitor Filanesib Enhances the Activity of Pomalidomide and Dexamethasone in Multiple Myeloma
Plasma Cell Disorders SUPPLEMENTARY APPENDIX The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma Susana Hernández-García, 1 Laura San-Segundo, 1 Lorena González-Méndez, 1 Luis A. Corchete, 1 Irena Misiewicz- Krzeminska, 1,2 Montserrat Martín-Sánchez, 1 Ana-Alicia López-Iglesias, 1 Esperanza Macarena Algarín, 1 Pedro Mogollón, 1 Andrea Díaz-Tejedor, 1 Teresa Paíno, 1 Brian Tunquist, 3 María-Victoria Mateos, 1 Norma C Gutiérrez, 1 Elena Díaz- Rodriguez, 1 Mercedes Garayoa 1* and Enrique M Ocio 1* 1Centro Investigación del Cáncer-IBMCC (CSIC-USAL) and Hospital Universitario-IBSAL, Salamanca, Spain; 2National Medicines Insti - tute, Warsaw, Poland and 3Array BioPharma, Boulder, Colorado, USA *MG and EMO contributed equally to this work ©2017 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2017.168666 Received: March 13, 2017. Accepted: August 29, 2017. Pre-published: August 31, 2017. Correspondence: [email protected] MATERIAL AND METHODS Reagents and drugs. Filanesib (F) was provided by Array BioPharma Inc. (Boulder, CO, USA). Thalidomide (T), lenalidomide (L) and pomalidomide (P) were purchased from Selleckchem (Houston, TX, USA), dexamethasone (D) from Sigma-Aldrich (St Louis, MO, USA) and bortezomib from LC Laboratories (Woburn, MA, USA). Generic chemicals were acquired from Sigma Chemical Co., Roche Biochemicals (Mannheim, Germany), Merck & Co., Inc. (Darmstadt, Germany). MM cell lines, patient samples and cultures. Origin, authentication and in vitro growth conditions of human MM cell lines have already been characterized (17, 18). The study of drug activity in the presence of IL-6, IGF-1 or in co-culture with primary bone marrow mesenchymal stromal cells (BMSCs) or the human mesenchymal stromal cell line (hMSC–TERT) was performed as described previously (19, 20). -

Supp Data.Pdf
Supplementary Methods Verhaak sub-classification The Verhaak called tumor subtype was assigned as determined in the Verhaak et al. manuscript, for those TCGA tumors that were included at the time. TCGA data for expression (Affymetrix platform, level 1) and methylation (Infinium platform level 3) were downloaded from the TCGA portal on 09/29/2011. Affymetrix data were processed using R and Biocondutor with a RMA algorithm using quantile normalization and custom CDF. Level 3 methylation data has already been processed and converted to a beta-value. The genes that make up the signature for each of the 4 Verhaak groups (i.e. Proneural, Neural, Mesenchymal, and Classical) were downloaded. For each tumor, the averaged expression of the 4 genes signatures was calculated generating 4 ‘metagene’ scores, one for each subtype, (Colman et al.2010) for every tumor. Then each subtype metagene score was z-score corrected to allow comparison among the 4 metagenes. Finally, for each tumor the subtype with the highest metagene z-score among the four was used to assign subtype. Thus by this method, a tumor is assigned to the group to which it has the strongest expression signature. The shortcoming is that if a tumor has a ‘dual personality’ – for instance has strong expression of both classical and mesenchymal signature genes, there is some arbitrariness to the tumor’s assignment. Western Blot Analysis and cell fractionation Western blot analysis was performed using standard protocols. To determine protein expression we used the following antibodies: TAZ (Sigma and BD Biosciences), YAP (Santa Cruz Biotechnology), CD44 (Cell Signaling), FN1 (BD Biosciences), TEAD1, TEAD2, TEAD3, TEAD4 (Santa Cruz Biotechnology), MST1, p-MST1, MOB1, LATS1, LATS2, 14-3-3-, 1 ACTG2, (Cell Signaling), p-LATS1/2 (Abcam), Flag (Sigma Aldrich), Actin (Calbiochem), CAV2 (BD Biosciences), CTGF (Santa Cruz), RUNX2 (Sigma Aldrich), Cylin A, Cyclin E, Cyclin B1, p-cdk1, p-cdk4 (Cell Signaling Technologies). -

Supplemental Table 1A. Differential Gene Expression Profile of Adehcd40l and Adehnull Treated Cells Vs Untreated Cells
Supplemental Table 1a. Differential Gene Expression Profile of AdEHCD40L and AdEHNull treated cells vs Untreated Cells Fold change Regulation Fold change Regulation ([AdEHCD40L] vs ([AdEHCD40L] ([AdEHNull] vs ([AdEHNull] vs Probe Set ID [Untreated]) vs [Untreated]) [Untreated]) [Untreated]) Gene Symbol Gene Title RefSeq Transcript ID NM_001039468 /// NM_001039469 /// NM_004954 /// 203942_s_at 2.02 down 1.00 down MARK2 MAP/microtubule affinity-regulating kinase 2 NM_017490 217985_s_at 2.09 down 1.00 down BAZ1A fibroblastbromodomain growth adjacent factor receptorto zinc finger 2 (bacteria-expressed domain, 1A kinase, keratinocyte NM_013448 /// NM_182648 growth factor receptor, craniofacial dysostosis 1, Crouzon syndrome, Pfeiffer 203638_s_at 2.10 down 1.01 down FGFR2 syndrome, Jackson-Weiss syndrome) NM_000141 /// NM_022970 1570445_a_at 2.07 down 1.01 down LOC643201 hypothetical protein LOC643201 XM_001716444 /// XM_001717933 /// XM_932161 231763_at 3.05 down 1.02 down POLR3A polymerase (RNA) III (DNA directed) polypeptide A, 155kDa NM_007055 1555368_x_at 2.08 down 1.04 down ZNF479 zinc finger protein 479 NM_033273 /// XM_001714591 /// XM_001719979 241627_x_at 2.15 down 1.05 down FLJ10357 hypothetical protein FLJ10357 NM_018071 223208_at 2.17 down 1.06 down KCTD10 potassium channel tetramerisation domain containing 10 NM_031954 219923_at 2.09 down 1.07 down TRIM45 tripartite motif-containing 45 NM_025188 242772_x_at 2.03 down 1.07 down Transcribed locus 233019_at 2.19 down 1.08 down CNOT7 CCR4-NOT transcription complex, subunit 7 NM_013354 -

Genetics of Abca4-Associated Diseases and Retinitis Pigmentosa
GENETICS OF ABCA4-ASSOCIATED DISEASES AND RETINITIS PIGMENTOSA Yajing (Angela) Xie Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 © 2016 Yajing (Angela) Xie All rights reserved ABSTRACT Genetics of ABCA4-Associated Diseases and Retinitis Pigmentosa Yajing (Angela) Xie Inherited retinal dystrophies encompass a broad group of genetic disorders affecting visual functions in as high as 1 in 3,000 individuals around the world. Common symptoms include loss of central, periphery, or night visions, and in severe cases progression to complete blindness. Syndromic forms also exist involving abnormalities in other parts of the body. Currently, more than 250 genes representing a wide variety of functional roles have been shown to be responsible for the disease phenotypes. Moreover, mutations in the same gene sometimes cause different phenotypes while mutations in multiple genes can give rise to the same clinical subtype, further demonstrating the level of complexity in these disorders. Such genetic heterogeneity has substantially complicated the process of pinpointing precise genetic causes underlying these conditions. The goal of my thesis research is to clarify the genetic causes underlying retinal dystrophies, with a primary focus on phenotypes resembling ABCA4-associated diseases and retinitis pigmentosa in both syndromic and non-syndromic forms. Recent advances in the next-generation sequencing (NGS), the high-throughput, ‘deep’ sequencing technology, have enabled several novel genes to be identified, or found new mutations in known genes. Nevertheless, a substantial fraction of unsolved cases still remain. The primary work in this thesis involves utilizing NGS, particularly whole- exome sequencing, to identify disease-causal mutations in families where at least one parent and affected or unaffected siblings are available. -
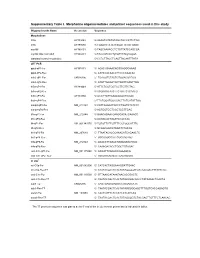
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Cell Division & Proliferation
ptglab.com 1 ANTIBODIES FOR CELL BIOLOGY CELL DIVISION & PROLIFERATION www.ptglab.com 2 Antibodies For Cell Biology: Cell Division & Proliferation Front & Back Cover: Immunohistochemistry of paraffin-embedded human skin tissue slide using Involucrin Antibody (55328-1-AP) at a dilution of 1:50 (10x objective). ptglab.com 3 WELCOME Foreword Cell division and proliferation are processes essential to life; they are processes that underpin growth and development and tissue maintenance. Cell division is the process by which cells must partition their DNA and cell contents appropriately into new daughter cells. This intricate mechanism enables cell proliferation, which must be tightly regulated in balance with cell loss through apoptosis or necrosis. These two processes can go wrong, leading to pathological proliferation, and in so doing form the basis of neoplastic disease. Throughout this catalog you will find hundreds The processes of cell division and proliferation of Proteintech®* antibodies relevant to cell require a wealth of proteins, as demonstrated division and proliferation. They cover mitosis by the long list of components above; if you and meiosis, DNA replication, transcription cannot identify an antibody for your target and translation, as well as protein expression, protein in these pages, please visit our website folding and trafficking and nutrient sensing at ptglab.com to search our full catalog of over and metabolism. There are also antibodies 10,000 pre-made antibodies. We’re confident against cell cycle control proteins featured; you will find the antibody you’re looking for. for instance you will find a poster dedicated to BRCA signaling, a pathway deregulated in many cancers.