A Bayesian Model for Inference of Metabolic Divergence Among Microbial Communities
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modeling and Computational Prediction of Metabolic Channelling
MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING by Christopher Morran Sanford A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Molecular Genetics University of Toronto © Copyright by Christopher Morran Sanford 2009 Abstract MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING Master of Science 2009 Christopher Morran Sanford Graduate Department of Molecular Genetics University of Toronto Metabolic channelling occurs when two enzymes that act on a common substrate pass that intermediate directly from one active site to the next without allowing it to diffuse into the surrounding aqueous medium. In this study, properties of channelling are investigated through the use of computational models and cell simulation tools. The effects of enzyme kinetics and thermodynamics on channelling are explored with the emphasis on validating the hypothesized roles of metabolic channelling in living cells. These simulations identify situations in which channelling can induce acceleration of reaction velocities and reduction in the free concentration of intermediate metabolites. Databases of biological information, including metabolic, thermodynamic, toxicity, inhibitory, gene fusion and physical protein interaction data are used to predict examples of potentially channelled enzyme pairs. The predictions are used both to support the hypothesized evolutionary motivations for channelling, and to propose potential enzyme interactions that may be worthy of future investigation. ii Acknowledgements I wish to thank my supervisor Dr. John Parkinson for the guidance he has provided during my time spent in his lab, as well as for his extensive help in the writing of this thesis. I am grateful for the advice of my committee members, Prof. -

ABSTRACT BOZDAG, AHMET. Investigation of Methanol and Formaldehyde Metabolism in Bacillus Methanolicus
ABSTRACT BOZDAG, AHMET. Investigation of Methanol and Formaldehyde Metabolism in Bacillus methanolicus.(Under the direction of Prof. Michael C. Flickinger). Bacillus methanolicus is a Gram-positive aerobic methylotroph growing optimally at 50-53 °C. Wild-type strains of B. methanolicus have been reported to secrete 58 g/l of L- glutamate in fed-batch cultures. Mutants of B. methanolicus created via classical mutangenesis can secrete 37 g/l of L-lysine, at 50 °C. The genes required for methylotrophyin B. methanolicus are encoded by an endogenous plasmid, pBM19 in strain MGA3, except for hexulose phosphate synthase (hps) and phosphohexuloisomerase (phi) which are encoded on the chromosome.It is a promising candidate for industrial production of chemical intermediates or amino acids from methanol. B. methanolicus employs the ribulose monophospate (RuMP) pathway to assimilate the carbon derived from the methanol, but enzymes that dissimilate carbon are not identified, although formaldehyde and formate were identified as intermediates by 13C NMR. It is important to understand how methanol is oxidized to formaldehyde and then, to formate and carbon dioxide. This study aims to elucidate the methanol dissimilation pathway of B. methanolicus. Growth rates of B. methanolicus MGA3 were assessed on methanol, mannitol, and glucose as a substrate. B. methanolicus achieved maximum growth rate, µmax, when growing on 25 mM methanol, 0.65±0.007 h-1, and it gradually decreased to 0.231±0.004 h-1 at 2Mmethanol concentration which demonstrates substrate inhibition. The maximum growth rates (µmax) of B. methanolicus MGA3 on mannitol and glucose are 0.532±0.002 and 0.336±0.003 h-1, respectively. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Prevalence of Campylobacter Jejuni in Small-Scale Broiler Operations and the Effects of Sulfadimethoxine Administered to Control C
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2014 Prevalence of Campylobacter jejuni in Small-Scale Broiler Operations and the Effects of Sulfadimethoxine Administered to Control C. jejuni Infection in Broilers Wannee Tangkham Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Life Sciences Commons Recommended Citation Tangkham, Wannee, "Prevalence of Campylobacter jejuni in Small-Scale Broiler Operations and the Effects of Sulfadimethoxine Administered to Control C. jejuni Infection in Broilers" (2014). LSU Doctoral Dissertations. 3200. https://digitalcommons.lsu.edu/gradschool_dissertations/3200 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. PREVALENCE OF CAMPYLOBACTER JEJUNI IN SMALL-SCALE BROILER OPERATIONS AND THE EFFECTS OF SULFADIMETHOXINE ADMINISTERED TO CONTROL C. JEJUNI INFECTION IN BROILERS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agriculture and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in School of Nutrition and Food Sciences by Wannee Tangkham B.S., Prince of Songkla University, 2004 M.S., McNeese State University, 2009 December 2014 ACKNOWLEDGEMENTS This major academic accomplishment in my life could not have been possible without so many people helping me and encouraging me. I would like to thank my major professor, Dr. Marlene Janes, for her guidance throughout my Ph.D. -

(12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel Et Al
USOO8561811 B2 (12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel et al. (45) Date of Patent: Oct. 22, 2013 (54) SUBSTRATE FOR IMMOBILIZING (56) References Cited FUNCTIONAL SUBSTANCES AND METHOD FOR PREPARING THE SAME U.S. PATENT DOCUMENTS 3,952,053 A 4, 1976 Brown, Jr. et al. (71) Applicants: Christian Gert Bluchel, Singapore 4.415,663 A 1 1/1983 Symon et al. (SG); Yanmei Wang, Singapore (SG) 4,576,928 A 3, 1986 Tani et al. 4.915,839 A 4, 1990 Marinaccio et al. (72) Inventors: Christian Gert Bluchel, Singapore 6,946,527 B2 9, 2005 Lemke et al. (SG); Yanmei Wang, Singapore (SG) FOREIGN PATENT DOCUMENTS (73) Assignee: Temasek Polytechnic, Singapore (SG) CN 101596422 A 12/2009 JP 2253813 A 10, 1990 (*) Notice: Subject to any disclaimer, the term of this JP 2258006 A 10, 1990 patent is extended or adjusted under 35 WO O2O2585 A2 1, 2002 U.S.C. 154(b) by 0 days. OTHER PUBLICATIONS (21) Appl. No.: 13/837,254 Inaternational Search Report for PCT/SG2011/000069 mailing date (22) Filed: Mar 15, 2013 of Apr. 12, 2011. Suen, Shing-Yi, et al. “Comparison of Ligand Density and Protein (65) Prior Publication Data Adsorption on Dye Affinity Membranes Using Difference Spacer Arms'. Separation Science and Technology, 35:1 (2000), pp. 69-87. US 2013/0210111A1 Aug. 15, 2013 Related U.S. Application Data Primary Examiner — Chester Barry (62) Division of application No. 13/580,055, filed as (74) Attorney, Agent, or Firm — Cantor Colburn LLP application No. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

The Complete Genome Sequence of the Gram-Positive Bacterium Bacillus Subtilis
articles The complete genome sequence of the Gram-positive bacterium Bacillus subtilis F. Kunst1, N. Ogasawara2, I. Moszer3, A. M. Albertini4, G. Alloni4, V. Azevedo5, M. G. Bertero3,4, P. Bessie` res5, A. Bolotin5, S. Borchert6, R. Borriss7, L. Boursier3, A. Brans8, M. Braun9, S. C. Brignell10,S.Bron11, S. Brouillet3,12, C. V. Bruschi13, B. Caldwell14, V. Capuano5, N. M. Carter10, S.-K. Choi15, J.-J. Codani16, I. F. Connerton17, N. J. Cummings17, R. A. Daniel18, F. Denizot19, K. M. Devine20,A.Du¨sterho¨ ft9, S. D. Ehrlich5, P.T. Emmerson21, K. D. Entian6, J. Errington18, C. Fabret19, E. Ferrari14, D. Foulger18, C. Fritz9, M. Fujita22, Y.Fujita23,S.Fuma24, A. Galizzi4, N. Galleron5, S.-Y.Ghim15, P.Glaser3, A. Goffeau25, E. J. Golightly26, G. Grandi27, G. Guiseppi19,B.J.Guy10, K. Haga28, J. Haiech19, C. R. Harwood10,A.He´naut29, H. Hilbert9, S. Holsappel11, S. Hosono30, M.-F. Hullo3, M. Itaya31, L. Jones32, B. Joris8, D. Karamata33, Y.Kasahara2, M. Klaerr-Blanchard3, C. Klein6, Y.Kobayashi30, P.Koetter6, G. Koningstein34, S. Krogh20, M. Kumano24, K. Kurita24, A. Lapidus5, S. Lardinois8, J. Lauber9, V. Lazarevic33, S.-M. Lee35, A. Levine36, H. Liu28, S. Masuda30, C. Maue¨ l33,C.Me´digue3,12, N. Medina36, R. P. Mellado37, M. Mizuno30, D. Moestl9, S. Nakai2, M. Noback11, D. Noone20, M. O’Reilly20, K. Ogawa24, A. Ogiwara38, B. Oudega34, S.-H. Park15, V. Parro37,T.M.Pohl39, D. Portetelle40, S. Porwollik7, A. M. Prescott18, E. Presecan3, P. Pujic5, B. Purnelle25, G. Rapoport1, M. Rey26, S. Reynolds33, M. Rieger41, C. Rivolta33, E. Rocha3,12,B.Roche36, M. -
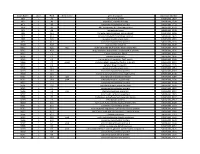
NTML 384-Well Array Well Identification
Strain Name plate Well Gene name gene discription Accession Number NE1 1 A1 cell surface protein SAUSA300_1327 NE2 1 A3 peptidase, rhomboid family SAUSA300_1509 NE3 1 A5 conserved hypothetical protein SAUSA300_1733 NE4 1 A7 ABC transporter ATP-binding protein SAUSA300_0309 NE5 1 A9 aminotransferase SAUSA300_2539 NE6 1 A11 formate dehydrogenase, alpha subunit SAUSA300_2258 NE7 1 A13 conserved hypothetical phage protein SAUSA300_1425 NE8 1 A15 putative membrane protein SAUSA300_1481 NE9 1 A17 conserved hypothetical protein SAUSA300_1294 NE10 1 A19 putative hemolysin III SAUSA300_2129 NE11 1 A21 recJ single-stranded-DNA-specific exonuclease RecJ SAUSA300_1592 NE12 1 A23 drug resistance transporter, EmrB/QacA subfamily SAUSA300_2126 NE13 1 C1 ribose transporter RbsU SAUSA300_0264 NE14 1 C3 putative transporter SAUSA300_2406 NE15 1 C5 transcriptional regulator, TetR family SAUSA300_2509 NE16 1 C7 moaD molybdopterin converting factor, subunit 1 SAUSA300_2221 NE17 1 C9 putative drug transporter SAUSA300_1705 NE18 1 C11 putative membrane protein SAUSA300_1851 NE19 1 C13 conserved hypothetical protein SAUSA300_0369 NE20 1 C15 transcriptional antiterminator, BglG family SAUSA300_0238 NE21 1 C17 upp uracil phosphoribosyltransferase SAUSA300_2066 NE22 1 C19 polA DNA polymerase I superfamily SAUSA300_1636 NE23 1 C21 conserved hypothetical protein SAUSA300_0740 NE24 1 C23 secretory antigen precursor SsaA SAUSA300_2503 NE25 1 E1 conserved hypothetical protein SAUSA300_1905 NE26 1 E3 coa staphylocoagulase precursor SAUSA300_0224 NE27 1 E5 nitric oxide -

1471-2164-6-174-S4.PDF (299.1Kb)
Sup_Table_2. Comparison of the whole genomes in Fig. 3A. Segment 1- Conserved in Bm, Bp, and Bt to Bp to Bt Gene Description % length % identity % length % identity BMA0001 chromosomal replication initiator protein DnaA 100 99 100 96 BMA0002 DNA polymerase III, beta subunit 100 100 100 99 BMA0003 DNA gyrase, B subunit 100 100 100 99 BMA0006 carboxymuconolactone decarboxylase family protein 100 98 100 99 BMA0010 hypothetical protein 100 99 100 92 BMA0011 hypothetical protein 100 100 100 91 BMA0014.1 hypothetical protein 100 99 96 94 BMA0018 hypothetical protein 100 99 100 95 BMA0019 FHA domain protein 100 100 100 94 BMA0020 protein kinase domain protein 100 99 100 90 BMA0023 conserved hypothetical protein 100 99 100 90 BMA0024 aldolase, class II 100 98 100 91 BMA0027 polysaccharide biosynthesis family protein 100 100 100 96 BMA0028 glycosyl transferase, group 1 family protein 100 99 100 94 BMA0029 mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase 100 99 100 92 BMA0030 ElaA family protein 100 99 100 90 BMA0032 glycosyl transferase, group 1 family protein 100 99 100 93 BMA0037 sigma-54 dependent transcriptional regulator 100 99 100 97 BMA0039 beta-mannosidase-related protein 100 99 100 91 BMA0040 conserved hypothetical protein 100 100 100 94 BMA0041 conserved hypothetical protein 100 99 100 95 BMA0042 acyl-CoA dehydrogenase domain protein 100 99 100 96 BMA0043 acyl carrier protein, putative 100 100 100 95 BMA0044 conserved hypothetical protein 100 99 100 96 BMA0045 conserved hypothetical protein 100 100 100 98 BMA0046 -

Proteomic Signatures of Bacillus Subtilis
1 Proteomic signatures of Bacillus subtilis Dissertation in fulfilment of the academic grade doctor rerum naturalium (Dr. rer. nat.) at the Faculty of Mathematics and Natural Sciences Ernst-Moritz-Arndt-University Greifswald Le Thi Tam born on 08.01.1979 in Bacninh, Vietnam Greifswald, Germany, 2006 2 Dekan: 1. Gutachter 1: 2. Gutachter 2: 3. Gutachter 3: Tag der Promotion: 3 Contents Contents 3 Abbreviations 5 Summary of thesis 7 Chapter 1: Introduction 9 1. Proteomic approachs and definition of proteomic signatures 10 2. B. subtilis as model organism for functional genomics 12 3. Proteome maps in Gram-positive bacteria 13 4. Regulation and function of stress responses 14 4.1. Heat shock response 14 4.2. Salt stress response 17 4.3. Oxidative stress response 19 4.4. Antibiotic response 21 5. Regulation and function of the starvation responses in B. subtilis 23 5.1. Glucose starvation response 24 5.2. Phosphate starvation response 26 5.3. Nitrogen starvation response 27 5.4. Tryptophan starvation response 29 5.5. The RelA-dependent stringent response 31 5.6. The CodY-dependent starvation response 32 5.7. The σH-dependent general starvation response 32 6. Degradation of aromatic compounds in microorganism 33 7. Scopes of thesis 36 Chapter 2: A comprehensive proteome map of growing Bacillus subtilis cells 39 Chapter 3: Proteome signatures for stress and starvation in Bacillus subtilis 69 as revealed by a 2D gel image color coding approach Chapter 4: Global gene expression profiling of Bacillus subtilis in response to 91 ammonium and tryptophan starvation as revealed by transcriptome and proteome analysis Chapter 5: Differential gene expression in response to phenol and catechol 131 reveal different metabolic activities for the degradation of aromatic compounds in Bacillus subtilis Chapter 6: Proteomic signature catalog of B. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Streptococcus Laboratory General Methods
The reference used for compiling the methods in Section I is: Murray, P.R., Baron, E. J., Jorgensen, J.J., Pfaller, M.A., and Yolken, R.H. Manual of Clinical Microbiology, 8th ed. ASM Press: Washington, DC, 2003. The Streptococcus species identification methods in Section II were compiled by Dr. Lynn Shewmaker. Also thanks to input from several individuals, including Richard Facklam and Lucia Teixeira. Section I. 1. Accuprobe-Enterococcus Test………….………..4 2. Accuprobe-Pneumococcus Test …………..….….4 3. Acid formation in carbohydrate broth..................5 4. Arginine Hydrolysis……………………….……….6 5. Bacitracin Test……………………………………..7 6. Bile-esculin Test…………………………………...8 7. Bile solubility Test …………………………………9 8. CAMP Test…......................................................10 9. Catalase Test......................................................11 10. Clindamycin test………………………………….12 11. Esculin hydrolysis……………………………….. 13 12. Gas from MRS broth……………………………...14 13. Gram Stain………………………………………...15 14. Growth at 10 & 45C……………………………. 17 15. Hemolysis………………………………………….18 16. Hippurate hydrolysis…………………………… 19 17. Lancefield Group Antigen………………………..20 18. Leucine amino peptidase (LAP)…………………21 19. Litmus Milk Test…………………………………..22 20. Motility………………………………………………23 21. 6.5% NaCl Tolerace Test...................................24 22. Optochin…………………………………………….25 23. Pigmentation....................................................... 26 24. Pyridoxal Requirement Test (Vitamin B6)……….27 25. Pyrrolidonlarylamindase (PYR)............................28 26.