RSC COFI Prelims 1..4
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Origin and Distribution of Phytophthora Cinnamomi
THE ORIGIN AND DISTRIBUTION OF PHYTOPHTHORA CINNAMOMI RANDS IN AUSTRALIAN NATIVE PLANT COMMUNITIES AND THE SIGNIFICANCE OF ITS ASSOCIATION WITH PARTICULAR PLANT SPECIES By B. H. PRATT* and W. A. HEATHER* [Manuscript received 23 October 1972] Abstract The origin, distribution, and disease association of P. cinnamomi in native plant communities in Australia has been examined. The fungus was isolated from the root zones of 31 plant genera in 16 families and is widespread throughout eastern and southern Australia and south-western Western Australia. Although the fungus is associated with disease in native plant communities it is also present in apparently non-diseased communities. Disease occurs usually only in environments disturbed by man, probably as a result of increase in population or activity of pre-existing fungal populations. The widespread distribution of P. cinnamomi in native vegetation in Australia, its occurrence in remote, undisturbed areas, and the apparent balance it has achieved with plant species of differing susceptibility to disease in some natural, undisturbed areas suggests that the fungus is likely to be indigenous to eastern Australia. Further, it may be partly responsible for the localized distribution of some plant species. I. INTRODUCTION The soil-borne fungus Phytophthora cinnamomi Rands is widely distributed in Australia, in horticultural plantings (Zentmyer and Thorn 1967; Pratt and Wrigley 1970; as well as personal communications from the New South Wales Department of Agriculture, Tasmanian Department of Agriculture, Victorian Department of Agriculture, and the Queensland Department of Primary Industries), and in conifer nurseries and plantings (Oxenham and Winks 1963; Bertus 1968; and personal communications from the New South Wales Forestry Commission, and the Queens land Department of Forestry). -

Biodiversity
Biodiversity KEY5 FACTS as hunting), as pasture grasses or as aquarium species Introduced (in the case of some marine species). They have also • Introduced species are been introduced accidentally, such as in shipments of recognised as a leading Species imported grain or in ballast water. cause of biodiversity loss Introduced plants, or weeds, can invade and world-wide. compete with native plant species for space, light, Trends water and nutrients and because of their rapid growth rates they can quickly smother native vegetation. • Rabbit numbers: a DECLINE since Similarly to weeds, many introduced animals compete introduction of Rabbit Haemorrhagic with and predate on native animals and impact on Disease (RHD, also known as calicivirus) native vegetation. They have high reproductive rates although the extent of the decline varies and can tolerate a wide range of habitats. As a result across the State. they often establish populations very quickly. •Fox numbers: DOWN in high priority Weeds can provide shelter for pest animals, conservation areas due to large-scale although they can provide food for or become habitat baiting programs; STILL A PROBLEM in for native animals. Blackberry, for example, is an ideal other parts of the State. habitat for the threatened Southern Brown Bandicoot. This illustrates the complexity of issues associated •Feral camel and deer numbers: UP. with pest control and highlights the need for control •Feral goat numbers: DECLINING across measures to have considered specific conservation Weed affected land – Mount Lofty Ranges the State. outcomes to be undertaken over time and to be Photo: Kym Nicolson •Feral pig numbers: UNKNOWN. -

Resistance of Castanea Clones to Phytophthora Cinnamomi: Testing and Genetic Control
Miranda-Fontaina et. al.·Silvae Genetica (2007) 56-1, 11-21 Resistance of Castanea Clones to Phytophthora Cinnamomi: Testing and Genetic Control By M. E. MIRANDA-FONTAÍÑA1), J. FERNÁNDEZ-LÓPEZ1), A. M. VETTRAINO2) and A. VANNINI2) (Received 23th May 2005) Summary Chestnut hybrid clone forest reproduction material for The resistance of chestnut clones to Phytophthora cin- commercial use in Spain must be approved as belonging namomi was evaluated by a soil inoculation experiment to qualified or controlled categories, and its value must under controlled environmental conditions, as well as by be demonstrated according to the requirements outlined excised and intact stem tests. One-year-old plants of in Directive 1999/105/EC (EUROPEAN COUNCIL, 1999) and fifty different clones were inoculated with two isolates of RD 289/2003 (BOLETIN OFICIAL DEL ESTADO, 2003). For Phytophthora cinnamomi and evaluated fourteen weeks successful wood production in Atlantic areas, stem qual- after inoculation. There were significant differences ity and growth as well as a certain level of resistance to among clones for all root and collar rot variables. There Phytophthora sp. are used as selection criteria. While were significant differences for isolates of P. cinnamomi the first two traits can be assessed in field trials, evalu- but only for the collar rot variables. A total of 84% of ation of resistance in terms of survival is not straightfor- plants grown in infested soil showed symptoms of root rot but only 50% of the plants with root rot, showed also ward, as it is difficult to differentiate the different fac- had collar rot. The roots of resistant clones were able to tors related to survival in the field. -
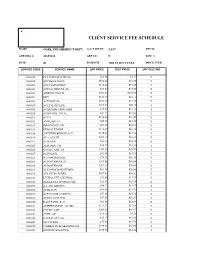
Oakland Sheriff's Dept Fee Schedule 02.09
CLIENT SERVICE FEE SCHEDULE NAME: OAKLAND SHERIFF'S DEPT ACCT SETUP: 2/1/87 REP ID: GRP BILL #: 22857610 GRP CD: N DISC %: FS ID: 01 FS DESCR: 2003 CLIENT FEES PRICE TYPE: SERVICE CODE SERVICE NAME UPF PRICE TEST PRICE UPF DISC IND 0000018 PLT SODIUM CITRATE $12.86 $4.37 Y 0000022 APC RESISTANCE $101.62 $34.55 Y 0000201 ACETAMINOPHEN $110.24 $37.48 Y 0000202 ACETALDEHYDE (B) $73.00 $73.00 N 0000203 ARSENIC,TISSUE $302.00 $302.00 N 0000204 DHT $182.70 $62.12 Y 0000205 ACETONE (B) $102.20 $34.75 Y 0000206 ACETYLCHOLINE $165.00 $56.10 Y 0000208 ACID PHOS, PROS, IMM $35.60 $12.10 Y 0000210 ACID PHOS, TOTAL $37.22 $12.65 Y 0000211 ACTH $110.00 $37.40 Y 0000212 AMYLASE (U) $45.45 $15.45 Y 0000213 IMMUNOFIX, UR $67.39 $22.91 Y 0000214 ETHOSUXIMIDE $112.07 $38.10 Y 0000216 ANTITHROMBIN III ACT $170.46 $57.96 Y 0000219 ALA, QUANT $107.10 $36.41 Y 0000223 ALBUMIN $22.88 $22.88 N 0000224 ALBUMIN, CSF $36.25 $12.33 Y 0000225 CYCLIC AMP, UR $205.80 $69.97 Y 0000227 ALDOLASE $32.08 $10.91 Y 0000228 A-2-MACROGLOB. $78.75 $26.78 Y 0000229 ALDOSTERONE (U) $251.58 $85.54 Y 0000230 ALDOSTERONE $207.10 $70.41 Y 0000231 ALK PHOS ISOENZYMES $61.34 $20.86 Y 0000232 AFP, FLUID W/RFX $107.40 $36.52 Y 0000233 LEUKOCYTE ALK PHOS $34.60 $11.76 Y 0000234 ALKALINE PHOSPHATASE $22.88 $22.88 N 0000235 A-1-ANTITRYPSN $40.22 $13.67 Y 0000236 AMIKACIN $99.91 $33.97 Y 0000237 AFP,TUMOR (CHIRON) $53.86 $18.31 Y 0000238 AMINO ACID SCR $87.15 $29.63 Y 0000240 RAST PANEL #103 $62.00 $62.00 N 0000241 AMPHETAMINE GC/MS $111.70 $37.98 Y 0000242 CYCLIC AMP $205.80 $69.97 Y 0000243 AMYLASE $16.42 $5.58 Y 0000248 HACKBERRY IGE $35.40 $35.40 N 0000249 ANA W/RFX $55.00 $18.70 Y 0000250 *AMIKACIN,PEAK&TROUGH $199.83 $67.94 Y 0000251 ANDROSTENEDIONE $180.65 $61.42 Y 0000252 ANTI-DIUR. -

Distribution of Methionine Sulfoxide Reductases in Fungi and Conservation of the Free- 2 Methionine-R-Sulfoxide Reductase in Multicellular Eukaryotes
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.26.433065; this version posted February 27, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Distribution of methionine sulfoxide reductases in fungi and conservation of the free- 2 methionine-R-sulfoxide reductase in multicellular eukaryotes 3 4 Hayat Hage1, Marie-Noëlle Rosso1, Lionel Tarrago1,* 5 6 From: 1Biodiversité et Biotechnologie Fongiques, UMR1163, INRAE, Aix Marseille Université, 7 Marseille, France. 8 *Correspondence: Lionel Tarrago ([email protected]) 9 10 Running title: Methionine sulfoxide reductases in fungi 11 12 Keywords: fungi, genome, horizontal gene transfer, methionine sulfoxide, methionine sulfoxide 13 reductase, protein oxidation, thiol oxidoreductase. 14 15 Highlights: 16 • Free and protein-bound methionine can be oxidized into methionine sulfoxide (MetO). 17 • Methionine sulfoxide reductases (Msr) reduce MetO in most organisms. 18 • Sequence characterization and phylogenomics revealed strong conservation of Msr in fungi. 19 • fRMsr is widely conserved in unicellular and multicellular fungi. 20 • Some msr genes were acquired from bacteria via horizontal gene transfers. 21 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.26.433065; this version posted February 27, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Tricholoma Aurantium
Tricholoma aurantium Pilzportrait Fungi, Dikarya, Basidiomycota, Agaricomycotina, Agaricomycetes, Agaricomycetidae, Agaricales, Tricholomataceae Tricholoma aurantium Orangeroter Ritterling Tricholoma aurantium Tricholoma aurantium (Schaeffer) Ricken 1915 Agaricus aurantia Schaeffer 1774 Agaricus aurantius Schaeffer 1774 Agaricus aurantius Schaeffer 1774 Amanita punctata var. aurantia (Schaeffer) Lamarck 1783 Amanita aurantia Lamarck 1783 Armillaria aurantia (Schaeffer) P. Kummer 1871 Gyrophila aurantia (Schaeffer) Quélet 1886 Mastoleucomyces aurantius (Schaeffer) Kuntze 1891 Melanoleuca aurantia (Schaeffer) Murrill 1914 Tricholoma aurantium (Schaeffer) Ricken 1915 makroskopisch Fruchtkörperfarbe / Farbspektrum Orange Fleischfarbe / Trama / Farbe Schnitt Fruchtkörper Weiss Hutfarbe Lebhaft orange - organgebraun Hutmerkmale Rand of etwas gerippt Stielmerkmale Genattert, mit Ring Lamellenmerkmale Im Alter an Schneiden fleckend, gekerbt Oxidation / Verfärbung: Fruchtkörper, Milch, Röhren Lamellen fleckend Sporenfarbe / Sporenpulver (Abwurf) Weiss olfaktorisch / organoleptisch Geruch / Geruchsprofil Stark nach Gurken Geschmack Zuerst nach Gurken, dann bitter und Bitterkeit ziemlich lang im Mund anhaltend, adstringierend botanisch / ökologisch Standort Picea, Kalkboden, collin bis alpin mikroskopisch Sporenmasse 4 x 5,5 µm - sehr kleine Sporen, teilweise fast rund Sporenmembran, Oberfläche, Skulptur Glatt Gattung/en: Tricholoma https://www.mycopedia.ch/pilze/1090.htm Siehe auch TRICHOLOMA_AURANTIUM www.mycopedia.ch - T. Flammer© 07.09.2021 -

A Bibliography of Mycology and Plant Pathology in Ireland, 1976 to 2000
Glasra 4: 119 – 188 (2008) A bibliography of mycology and plant pathology in Ireland, 1976 to 2000 A. MANGAN 24 Treesdale, Blackrock, Co. Dublin ABSTRACT This bibliography includes medical and veterinary mycology, and plant pathology relating to bacteriology and virology. It is based on published work from Ireland, between 1976 and 2000 inclusive, and comprises 859 references. An index of topics is included. There is also a list of 193 theses for which Master or Doctorate degrees were awarded. INTRODUCTION In his publication, Mycology and Plant Pathology in Ireland, Muskett (1976) gave an account of the history and development of mycology and plant pathology in Ireland since the first traceable mention of Irish fungi in the scientific sense in 1726. His work included a bibliography containing 1,355 titles, in chronological order, covering the period from 1726 to 1975. The bibliography presented here covers the years 1976 to 2000. The basis of the present work was to search for work on mycology and plant pathology published by authors with an Irish address. Where work relating to Ireland, which was published from an address abroad, was noted, it is included but it was not possible to make a full search for such work. The principal sources for this work were: The Review of Plant Pathology and the Review of Medical and Veterinary Mycology, both published by CAB International. The following journals were also checked for relevant material: Irish Naturalists’ Journal Proceedings of the Royal Irish Academy Proceedings of meetings published by the Royal Irish Academy Scientific Proceedings of the Royal Dublin Society Journal of Life Sciences of the Royal Dublin Society Irish Journal of Agricultural Research later named the Irish Journal of Agricultural and Food Research Mycologist Field Mycology Various reports of meetings, institutes and societies exist, but are not included as they are not readily available in libraries. -

A Nomenclatural Study of Armillaria and Armillariella Species
A Nomenclatural Study of Armillaria and Armillariella species (Basidiomycotina, Tricholomataceae) by Thomas J. Volk & Harold H. Burdsall, Jr. Synopsis Fungorum 8 Fungiflora - Oslo - Norway A Nomenclatural Study of Armillaria and Armillariella species (Basidiomycotina, Tricholomataceae) by Thomas J. Volk & Harold H. Burdsall, Jr. Printed in Eko-trykk A/S, Førde, Norway Printing date: 1. August 1995 ISBN 82-90724-14-4 ISSN 0802-4966 A Nomenclatural Study of Armillaria and Armillariella species (Basidiomycotina, Tricholomataceae) by Thomas J. Volk & Harold H. Burdsall, Jr. Synopsis Fungorum 8 Fungiflora - Oslo - Norway 6 Authors address: Center for Forest Mycology Research Forest Products Laboratory United States Department of Agriculture Forest Service One Gifford Pinchot Dr. Madison, WI 53705 USA ABSTRACT Once a taxonomic refugium for nearly any white-spored agaric with an annulus and attached gills, the concept of the genus Armillaria has been clarified with the neotypification of Armillaria mellea (Vahl:Fr.) Kummer and its acceptance as type species of Armillaria (Fr.:Fr.) Staude. Due to recognition of different type species over the years and an extremely variable generic concept, at least 274 species and varieties have been placed in Armillaria (or in Armillariella Karst., its obligate synonym). Only about forty species belong in the genus Armillaria sensu stricto, while the rest can be placed in forty-three other modem genera. This study is based on original descriptions in the literature, as well as studies of type specimens and generic and species concepts by other authors. This publication consists of an alphabetical listing of all epithets used in Armillaria or Armillariella, with their basionyms, currently accepted names, and other obligate and facultative synonyms. -

No.124, Year 2013
МАТИЦА СРПСКА ОДЕЉЕЊЕ ЗА ПРИРОДНЕ НАУКЕ ЗБОРНИК МАТИЦЕ СРПСКЕ ЗА ПРИРОДНЕ НАУКЕ MATICA SRPSKA DEPARTMENT OF NATURAL SCIENCES JOURNAL FOR NATURAL SCIENCES Покренут 1951 / First published in 1951. Published as Научни зборник, серија природних наука until the tenth issue (1955), as the Series for Natural Science from the eleventh issue (1956) — Зборник за pриродне науке, and under its present title since the sixty-sixth issue (1984) Главни уредници / Editors-in-Chief Miloš Jovanović (1951), Branislav Bukurov (1952—1969), Lazar Stojković (1970—1976), Slobodan Glumac (1977—1996), Rudolf Kastori (1996—2012), Ivana Maksimović (2013—) 124 Уредништво / Editorial Board Consulting Editors S. GAJIN A. ATANASSOV, Bulgaria S. ĆURČIĆ P. HOCKING, Australia V. JANJIĆ A. RODZKIN, Belarus V. JOVIĆ K. ROUBELAKIS-ANGELAKIS, Greece D. KAPOR S. STOJILJKOVIĆ, USA R. KASTORI G. SCHILING, Germany L. LEPŠANOVIĆ GY. VÁRALLYAY, Hungary I. MAKSIMOVIĆ A. VENEZIA, Italy V. MARIĆ M. ŠKRINJAR Articals are available in full-text at the web site of Matica Srpska and in the fol- lowing data bases: Serbian Citation Index, EBSCO Academic Search Complet, abstract level at Agris (FAO), CAB Abstracts, CABI Full-Text and Thomson Reuters Master Journal List. Главни и одговорни уредник / Editor-in-Chief IVANA MAKSIMOVIĆ YU ISSN 0352-4906 UDK 5/6 (05) MATICA SRPSKA JOURNAL FOR NATURAL SCIENCES 124 NOVI SAD 2013 SCIENTIFIC BOARD 5TH INTERNATIONAL SCIENTIFIC MEETING MYCOLOGY, MYCOTOXICOLOGY AND MYCOSES Professor Dragan Stanić, President of Matica Srpska Professor Ferenc Balaž, Ph.D. Professor Dilek Heperkan, Ph.D. Akademician Dragutin Đukić Professor Hans van Egmond, Ph.D. Professor Elias Nerantzis, Ph.D. Akademician Rudolf Kastori Professor Đoko Kungulovski, Ph.D. -

Checklist of the Species of the Genus Tricholoma (Agaricales, Agaricomycetes) in Estonia
Folia Cryptog. Estonica, Fasc. 47: 27–36 (2010) Checklist of the species of the genus Tricholoma (Agaricales, Agaricomycetes) in Estonia Kuulo Kalamees Institute of Ecology and Earth Sciences, University of Tartu, 40 Lai St. 51005, Tartu, Estonia. Institute of Agricultural and Environmental Sciences, Estonian University of Life Sciences, 181 Riia St., 51014 Tartu, Estonia E-mail: [email protected] Abstract: 42 species of genus Tricholoma (Agaricales, Agaricomycetes) have been recorded in Estonia. A checklist of these species with ecological, phenological and distribution data is presented. Kokkukvõte: Perekonna Tricholoma (Agaricales, Agaricomycetes) liigid Eestis Esitatakse kriitiline nimestik koos ökoloogiliste, fenoloogiliste ja levikuliste andmetega heiniku perekonna (Tricholoma) 42 liigi (Agaricales, Agaricomycetes) kohta Eestis. INTRODUCTION The present checklist contains 42 Tricholoma This checklist also provides data on the ecol- species recorded in Estonia. All the species in- ogy, phenology and occurrence of the species cluded (except T. gausapatum) correspond to the in Estonia (see also Kalamees, 1980a, 1980b, species conceptions established by Christensen 1982, 2000, 2001b, Kalamees & Liiv, 2005, and Heilmann-Clausen (2008) and have been 2008). The following data are presented on each proved by relevant exsiccates in the mycothecas taxon: (1) the Latin name with a reference to the TAAM of the Institute of Agricultural and Envi- initial source; (2) most important synonyms; (3) ronmental Sciences of the Estonian University reference to most important and representative of Life Sciences or TU of the Natural History pictures (iconography) in the mycological litera- Museum of the Tartu University. In this paper ture used in identifying Estonian species; (4) T. gausapatum is understand in accordance with data on the ecology, phenology and distribution; Huijsman, 1968 and Bon, 1991. -

Endophytic Fungi: Treasure for Anti-Cancerous Compounds
International Journal of Pharmacy and Pharmaceutical Sciences ISSN- 0975-1491 Vol 8, Issue 8, 2016 Review Article ENDOPHYTIC FUNGI: TREASURE FOR ANTI-CANCEROUS COMPOUNDS ANAND DILIP FIRODIYAa*, RAJESH KUMAR TENGURIAb aCSRD, Peoples University, Bhopal 462037, Madhya Pradesh, India, bDepartment of Botany, Govt. PG College, Rajgarh 496551, Madhya Pradesh, India Email: [email protected] Received: 22 Apr 2016 Revised and Accepted: 20 June 2016 ABSTRACT Endophytic fungi that live asymptomatically inside the plant tissues have novel bioactive metabolites exhibiting a variety of biological activities, especially against cancer. This review highlights the research progress on the production of anticancer compounds by endophytic fungi from 1990- 2015. Anticancer activity is generally associated with the cytotoxicity of the compounds present in the endophytic fungi. The ubiquitous nature of endophytic fungi synthesise diverse chemicals with promising anticancer activity from either their original host or related species. Modification in fermentation parameters and genetic insight of endophytes may produce novel anti-cancerous compounds. Keywords: Cancer, Medicinal plants, Secondary metabolites © 2016 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/) INTRODUCTION endophytic fungi detectable by high-performance liquid chromate- graphy, nuclear magnetic resonance, mass spectrophotometer and The interest in the biogenic medicines has revived throughout the X-ray crystallography and its cytotoxicity of the bioactive world, as the increase in awareness of the health hazards and compounds against cancer cell lines. The compounds with potential toxicity associated with the random use of synthetic drugs and application were also considered in the selection of antitumor antibiotics [1]. -

Pipestem Foray Overview
Volume 49:1 January ⁄ February 2008 www.namyco.org Pipestem Foray Overview An East-Coaster’s Perspective A West-Coaster’s Perspective by Dave Wasilewski by Debbie Viess For about 25 years now I have As Steve Trudell rightly pointed out hunted and studied wild mush- to me, don’t gloat about your mush- rooms, but I’ve never been active in rooms until they are safely in your a club. The NAMA Orson K. Miller basket! The continuing “Curse of Memorial Foray held in Pipestem, NAMA” (some call it global warm- WV, this past August was the first ing) slipped in the back door, behind such event that I have ever at- the earlier and heartening West tended. Virginia thunderstorms. Extreme I must admit that, as I drove heat and lack of rain for the previ- south on Interstate 81 through two ous couple of weeks made condi- solid hours of Pennsylvania rainfall tions on the ground challenging for on an eight-hour trip to a place hopeful finders of fungi. Chlorosplenium aeruginascens, one of where little or no rain had fallen for Luckily, my Southern Belle the many delights found at Pipestem. over a week, for the purpose of hostess with the mostest, Coleman hunting wild mushrooms, I felt a bit McCleneghan, took me on a few names like Gyroporus and Pulvero- conflicted. My mind wandered pre-NAMA forays in Virginia, where boletus, tucked among the through conifer groves in the conditions were much improved. My many shades of forest green and Poconos where imaginary boletes very first walk ever along the brown.