4766.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Targeting Hedgehog Signalling Through the Ubiquitylation Process: the Multiple Roles of the HECT-E3 Ligase Itch
cells Review Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch Paola Infante 1,†, Ludovica Lospinoso Severini 2,† , Flavia Bernardi 2, Francesca Bufalieri 2 and Lucia Di Marcotullio 2,3,* 1 Center for Life NanoScience@Sapienza, Istituto Italiano di Tecnologia, 00161 Rome, Italy; [email protected] 2 Department of Molecular Medicine, University of Rome La Sapienza, 00161 Rome, Italy; [email protected] (L.L.S.); fl[email protected] (F.B.); [email protected] (F.B.) 3 Istituto Pasteur-Fondazione Cenci Bolognetti, University of Rome La Sapienza, 00161 Rome, Italy * Correspondence: [email protected]; Tel.: +39-06-49255657 † These authors contributed equally to this work. Received: 28 December 2018; Accepted: 26 January 2019; Published: 29 January 2019 Abstract: Hedgehog signalling (Hh) is a developmental conserved pathway strongly involved in cancers when deregulated. This important pathway is orchestrated by numerous regulators, transduces through distinct routes and is finely tuned at multiple levels. In this regard, ubiquitylation processes stand as essential for controlling Hh pathway output. Although this post-translational modification governs proteins turnover, it is also implicated in non-proteolytic events, thereby regulating the most important cellular functions. The HECT E3 ligase Itch, well known to control immune response, is emerging to have a pivotal role in tumorigenesis. By illustrating Itch specificities on Hh signalling key components, here we review the role of this HECT E3 ubiquitin ligase in suppressing Hh-dependent tumours and explore its potential as promising target for innovative therapeutic approaches. Keywords: Hedgehog; cancer; ubiquitylation; Itch; Numb; β-arrestin2; GLI1; SuFu; Patched1 1. -
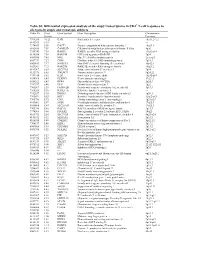
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -
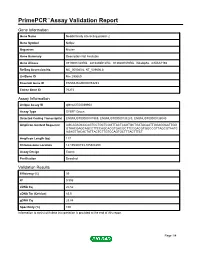
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Nedd4 family interacting protein 2 Gene Symbol Ndfip2 Organism Mouse Gene Summary Description Not Available Gene Aliases 0710001O20Rik, 2810436B12Rik, 9130207N19Rik, N4wbp5a, mKIAA1165 RefSeq Accession No. NC_000080.6, NT_039606.8 UniGene ID Mm.290669 Ensembl Gene ID ENSMUSG00000053253 Entrez Gene ID 76273 Assay Information Unique Assay ID qMmuCED0039954 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000181969, ENSMUST00000138283, ENSMUST00000136040 Amplicon Context Sequence AGCAGAGCCCAGTCCTGCTCGGTTACTCAGTGCTGATACAATTGGAGGAATTGG GTAACGAGCAGCCTTCCAGCACGTGACGCTTCCGACGTGGCCGTTAGCGTAATC AGAGTTACACTATTACTCTTGTCCAGTGCTTTACTTTCT Amplicon Length (bp) 117 Chromosome Location 14:105308153-105308299 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 99 R2 0.996 cDNA Cq 20.52 cDNA Tm (Celsius) 83.5 gDNA Cq 23.84 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Ndfip2, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Variation in Genomic Landscape of Clear Cell Renal Cell Carcinoma
ARTICLE Received 30 May 2014 | Accepted 3 Sep 2014 | Published 29 Oct 2014 DOI: 10.1038/ncomms6135 Variation in genomic landscape of clear cell renal cell carcinoma across Europe Ghislaine Scelo1,*, Yasser Riazalhosseini2,3,*, Liliana Greger4,*, Louis Letourneau3,*, Mar Gonza`lez-Porta4,*, Magdalena B. Wozniak1, Mathieu Bourgey3, Patricia Harnden5, Lars Egevad6, Sharon M. Jackson5, Mehran Karimzadeh2,3, Madeleine Arseneault2,3, Pierre Lepage3, Alexandre How-Kit7, Antoine Daunay7, Victor Renault7,He´le`ne Blanche´7, Emmanuel Tubacher7, Jeremy Sehmoun7, Juris Viksna8, Edgars Celms8, Martins Opmanis8, Andris Zarins8, Naveen S. Vasudev5, Morag Seywright9, Behnoush Abedi-Ardekani1, Christine Carreira1, Peter J. Selby5, Jon J. Cartledge10, Graham Byrnes1, Jiri Zavadil1, Jing Su4, Ivana Holcatova11, Antonin Brisuda12, David Zaridze13, Anush Moukeria13, Lenka Foretova14, Marie Navratilova14, Dana Mates15, Viorel Jinga16, Artem Artemov17, Artem Nedoluzhko18, Alexander Mazur17, Sergey Rastorguev18, Eugenia Boulygina18, Simon Heath19, Marta Gut19, Marie-Therese Bihoreau20, Doris Lechner20, Mario Foglio20, Ivo G. Gut19, Konstantin Skryabin17,18, Egor Prokhortchouk17,18, Anne Cambon-Thomsen21, Johan Rung4, Guillaume Bourque2,3, Paul Brennan1,Jo¨rg Tost20, Rosamonde E. Banks5,AlvisBrazma4 & G. Mark Lathrop2,3,7,20,w The incidence of renal cell carcinoma (RCC) is increasing worldwide, and its prevalence is particularly high in some parts of Central Europe. Here we undertake whole-genome and transcriptome sequencing of clear cell RCC (ccRCC), the most common form of the disease, in patients from four different European countries with contrasting disease incidence to explore the underlying genomic architecture of RCC. Our findings support previous reports on frequent aberrations in the epigenetic machinery and PI3K/mTOR signalling, and uncover novel pathways and genes affected by recurrent mutations and abnormal transcriptome patterns including focal adhesion, components of extracellular matrix (ECM) and genes encoding FAT cadherins. -

Supplementary Data
Supplemental figures Supplemental figure 1: Tumor sample selection. A total of 98 thymic tumor specimens were stored in Memorial Sloan-Kettering Cancer Center tumor banks during the study period. 64 cases corresponded to previously untreated tumors, which were resected upfront after diagnosis. Adjuvant treatment was delivered in 7 patients (radiotherapy in 4 cases, cyclophosphamide- doxorubicin-vincristine (CAV) chemotherapy in 3 cases). 34 tumors were resected after induction treatment, consisting of chemotherapy in 16 patients (cyclophosphamide-doxorubicin- cisplatin (CAP) in 11 cases, cisplatin-etoposide (PE) in 3 cases, cisplatin-etoposide-ifosfamide (VIP) in 1 case, and cisplatin-docetaxel in 1 case), in radiotherapy (45 Gy) in 1 patient, and in sequential chemoradiation (CAP followed by a 45 Gy-radiotherapy) in 1 patient. Among these 34 patients, 6 received adjuvant radiotherapy. 1 Supplemental Figure 2: Amino acid alignments of KIT H697 in the human protein and related orthologs, using (A) the Homologene database (exons 14 and 15), and (B) the UCSC Genome Browser database (exon 14). Residue H697 is highlighted with red boxes. Both alignments indicate that residue H697 is highly conserved. 2 Supplemental Figure 3: Direct comparison of the genomic profiles of thymic squamous cell carcinomas (n=7) and lung primary squamous cell carcinomas (n=6). (A) Unsupervised clustering analysis. Gains are indicated in red, and losses in green, by genomic position along the 22 chromosomes. (B) Genomic profiles and recurrent copy number alterations in thymic carcinomas and lung squamous cell carcinomas. Gains are indicated in red, and losses in blue. 3 Supplemental Methods Mutational profiling The exonic regions of interest (NCBI Human Genome Build 36.1) were broken into amplicons of 500 bp or less, and specific primers were designed using Primer 3 (on the World Wide Web for general users and for biologist programmers (see Supplemental Table 2) [1]. -

Supplementary Table S1. List of Differentially Expressed
Supplementary table S1. List of differentially expressed transcripts (FDR adjusted p‐value < 0.05 and −1.4 ≤ FC ≥1.4). 1 ID Symbol Entrez Gene Name Adj. p‐Value Log2 FC 214895_s_at ADAM10 ADAM metallopeptidase domain 10 3,11E‐05 −1,400 205997_at ADAM28 ADAM metallopeptidase domain 28 6,57E‐05 −1,400 220606_s_at ADPRM ADP‐ribose/CDP‐alcohol diphosphatase, manganese dependent 6,50E‐06 −1,430 217410_at AGRN agrin 2,34E‐10 1,420 212980_at AHSA2P activator of HSP90 ATPase homolog 2, pseudogene 6,44E‐06 −1,920 219672_at AHSP alpha hemoglobin stabilizing protein 7,27E‐05 2,330 aminoacyl tRNA synthetase complex interacting multifunctional 202541_at AIMP1 4,91E‐06 −1,830 protein 1 210269_s_at AKAP17A A‐kinase anchoring protein 17A 2,64E‐10 −1,560 211560_s_at ALAS2 5ʹ‐aminolevulinate synthase 2 4,28E‐06 3,560 212224_at ALDH1A1 aldehyde dehydrogenase 1 family member A1 8,93E‐04 −1,400 205583_s_at ALG13 ALG13 UDP‐N‐acetylglucosaminyltransferase subunit 9,50E‐07 −1,430 207206_s_at ALOX12 arachidonate 12‐lipoxygenase, 12S type 4,76E‐05 1,630 AMY1C (includes 208498_s_at amylase alpha 1C 3,83E‐05 −1,700 others) 201043_s_at ANP32A acidic nuclear phosphoprotein 32 family member A 5,61E‐09 −1,760 202888_s_at ANPEP alanyl aminopeptidase, membrane 7,40E‐04 −1,600 221013_s_at APOL2 apolipoprotein L2 6,57E‐11 1,600 219094_at ARMC8 armadillo repeat containing 8 3,47E‐08 −1,710 207798_s_at ATXN2L ataxin 2 like 2,16E‐07 −1,410 215990_s_at BCL6 BCL6 transcription repressor 1,74E‐07 −1,700 200776_s_at BZW1 basic leucine zipper and W2 domains 1 1,09E‐06 −1,570 222309_at -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. -

NDFIP1 Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap13449c
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 NDFIP1 Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP13449c Specification NDFIP1 Antibody (Center) - Product Information Application WB,E Primary Accession Q9BT67 Other Accession Q5U2S1, Q8R0W6, NP_085048.1 Reactivity Human Predicted Mouse, Rat Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Calculated MW 24899 Antigen Region 77-106 NDFIP1 Antibody (Center) - Additional Information Gene ID 80762 Anti-NDFIP1 Antibody (Center) at 1:1000 dilution + PC-3 whole cell lysate Other Names Lysates/proteins at 20 µg per lane. NEDD4 family-interacting protein 1, Breast Secondary Goat Anti-Rabbit IgG, (H+L), cancer-associated protein SGA-1M, NEDD4 Peroxidase conjugated at 1/10000 dilution. WW domain-binding protein 5, Putative Predicted band size : 25 kDa MAPK-activating protein PM13, Putative NF-kappa-B-activating protein 164, Putative Blocking/Dilution buffer: 5% NFDM/TBST. NFKB and MAPK-activating protein, NDFIP1, N4WBP5 NDFIP1 Antibody (Center) - Background Target/Specificity This NDFIP1 antibody is generated from The protein encoded by this gene belongs to a rabbits immunized with a KLH conjugated small group of evolutionarily conserved synthetic peptide between 77-106 amino proteins with three transmembrane domains. It acids from the Central region of human is a potential target for ubiquitination by the NDFIP1. Nedd4 family of proteins. This protein is thought to be part of a family of integral Golgi Dilution membrane proteins. WB~~1:1000 NDFIP1 Antibody (Center) - References Format Purified polyclonal antibody supplied in PBS Mund, T., et al. Proc. Natl. Acad. Sci. U.S.A. -

Functional Genomic Annotation of Genetic Risk Loci Highlights Inflammation and Epithelial Biology Networks in CKD
BASIC RESEARCH www.jasn.org Functional Genomic Annotation of Genetic Risk Loci Highlights Inflammation and Epithelial Biology Networks in CKD Nora Ledo, Yi-An Ko, Ae-Seo Deok Park, Hyun-Mi Kang, Sang-Youb Han, Peter Choi, and Katalin Susztak Renal Electrolyte and Hypertension Division, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania ABSTRACT Genome-wide association studies (GWASs) have identified multiple loci associated with the risk of CKD. Almost all risk variants are localized to the noncoding region of the genome; therefore, the role of these variants in CKD development is largely unknown. We hypothesized that polymorphisms alter transcription factor binding, thereby influencing the expression of nearby genes. Here, we examined the regulation of transcripts in the vicinity of CKD-associated polymorphisms in control and diseased human kidney samples and used systems biology approaches to identify potentially causal genes for prioritization. We interro- gated the expression and regulation of 226 transcripts in the vicinity of 44 single nucleotide polymorphisms using RNA sequencing and gene expression arrays from 95 microdissected control and diseased tubule samples and 51 glomerular samples. Gene expression analysis from 41 tubule samples served for external validation. 92 transcripts in the tubule compartment and 34 transcripts in glomeruli showed statistically significant correlation with eGFR. Many novel genes, including ACSM2A/2B, FAM47E, and PLXDC1, were identified. We observed that the expression of multiple genes in the vicinity of any single CKD risk allele correlated with renal function, potentially indicating that genetic variants influence multiple transcripts. Network analysis of GFR-correlating transcripts highlighted two major clusters; a positive correlation with epithelial and vascular functions and an inverse correlation with inflammatory gene cluster. -

Gene-Expression and in Vitro Function of Mesenchymal Stromal Cells Are Affected in Juvenile Myelomonocytic Leukemia
Myeloproliferative Disorders SUPPLEMENTARY APPENDIX Gene-expression and in vitro function of mesenchymal stromal cells are affected in juvenile myelomonocytic leukemia Friso G.J. Calkoen, 1 Carly Vervat, 1 Else Eising, 2 Lisanne S. Vijfhuizen, 2 Peter-Bram A.C. ‘t Hoen, 2 Marry M. van den Heuvel-Eibrink, 3,4 R. Maarten Egeler, 1,5 Maarten J.D. van Tol, 1 and Lynne M. Ball 1 1Department of Pediatrics, Immunology, Hematology/Oncology and Hematopoietic Stem Cell Transplantation, Leiden University Med - ical Center, the Netherlands; 2Department of Human Genetics, Leiden University Medical Center, Leiden, the Netherlands; 3Dutch Childhood Oncology Group (DCOG), The Hague, the Netherlands; 4Princess Maxima Center for Pediatric Oncology, Utrecht, the Nether - lands; and 5Department of Hematology/Oncology and Hematopoietic Stem Cell Transplantation, Hospital for Sick Children, University of Toronto, ON, Canada ©2015 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.126938 Manuscript received on March 5, 2015. Manuscript accepted on August 17, 2015. Correspondence: [email protected] Supplementary data: Methods for online publication Patients Children referred to our center for HSCT were included in this study according to a protocol approved by the institutional review board (P08.001). Bone-marrow of 9 children with JMML was collected prior to treatment initiation. In addition, bone-marrow after HSCT was collected from 5 of these 9 children. The patients were classified following the criteria described by Loh et al.(1) Bone-marrow samples were sent to the JMML-reference center in Freiburg, Germany for genetic analysis. Bone-marrow samples of healthy pediatric hematopoietic stem cell donors (n=10) were used as control group (HC).