Uncovering Potential Roles of Differentially Expressed Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

A Flexible Microfluidic System for Single-Cell Transcriptome Profiling
www.nature.com/scientificreports OPEN A fexible microfuidic system for single‑cell transcriptome profling elucidates phased transcriptional regulators of cell cycle Karen Davey1,7, Daniel Wong2,7, Filip Konopacki2, Eugene Kwa1, Tony Ly3, Heike Fiegler2 & Christopher R. Sibley 1,4,5,6* Single cell transcriptome profling has emerged as a breakthrough technology for the high‑resolution understanding of complex cellular systems. Here we report a fexible, cost‑efective and user‑ friendly droplet‑based microfuidics system, called the Nadia Instrument, that can allow 3′ mRNA capture of ~ 50,000 single cells or individual nuclei in a single run. The precise pressure‑based system demonstrates highly reproducible droplet size, low doublet rates and high mRNA capture efciencies that compare favorably in the feld. Moreover, when combined with the Nadia Innovate, the system can be transformed into an adaptable setup that enables use of diferent bufers and barcoded bead confgurations to facilitate diverse applications. Finally, by 3′ mRNA profling asynchronous human and mouse cells at diferent phases of the cell cycle, we demonstrate the system’s ability to readily distinguish distinct cell populations and infer underlying transcriptional regulatory networks. Notably this provided supportive evidence for multiple transcription factors that had little or no known link to the cell cycle (e.g. DRAP1, ZKSCAN1 and CEBPZ). In summary, the Nadia platform represents a promising and fexible technology for future transcriptomic studies, and other related applications, at cell resolution. Single cell transcriptome profling has recently emerged as a breakthrough technology for understanding how cellular heterogeneity contributes to complex biological systems. Indeed, cultured cells, microorganisms, biopsies, blood and other tissues can be rapidly profled for quantifcation of gene expression at cell resolution. -

Distinct Roles of Jun : Fos and Jun : ATF Dimers in Oncogenesis
Oncogene (2001) 20, 2453 ± 2464 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis Hans van Dam*,1 and Marc Castellazzi2 1Department of Molecular Cell Biology, Leiden University Medical Center, Sylvius Laboratories, PO Box 9503, 2300 RA Leiden, The Netherlands; 2Unite de Virologie Humaine, Institut National de la Sante et de la Recherche MeÂdicale (INSERM-U412), Ecole Normale SupeÂrieure, 46 alleÂe d'Italie, 69364 Lyon Cedex 07, France Jun : Fos and Jun : ATF complexes represent two classes dimers with emphasis on their roles in oncogenic of AP-1 dimers that (1) preferentially bind to either transformation in avian model systems. Previous heptameric or octameric AP-1 binding sites, and (2) are reviews on AP-1 and cell transformation include dierently regulated by cellular signaling pathways and references: (Angel and Karin, 1991; Wisdom, 1999; oncogene products. To discriminate between the func- Vogt, 1994; Karin et al., 1997; van Dam and van der tions of Jun : Fos, Jun: ATF and Jun : Jun, mutants were Eb, 1994; Hagmeyer et al., 1995). developed that restrict the ability of Jun to dimerize either to itself, or to Fos(-like) or ATF(-like) partners. Introduction of these mutants in chicken embryo Jun : Fos and Jun : ATF transcription factors: dimeric ®broblasts shows that Jun : Fra2 and Jun : ATF2 dimers complexes with variable composition and activities play distinct, complementary roles in in vitro oncogenesis by inducing either anchorage independence or growth AP-1 sub-units: members of the bZip protein family factor independence, respectively. -

The EWS/ATF1 Fusion Protein Contains a Dispersed Activation Domain That Functions Directly
Oncogene (1998) 16, 1625 ± 1631 1998 Stockton Press All rights reserved 0950 ± 9232/98 $12.00 The EWS/ATF1 fusion protein contains a dispersed activation domain that functions directly Shu Pan, Koh Yee Ming, Theresa A Dunn, Kim KC Li and Kevin AW Lee Department of Biology, Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, P.R.C. Naturally occurring chromosomal fusion of the Ewings 1994). For all of the above malignancies, the EWS Sarcoma Oncogene (EWS) to distinct cellular transcrip- fusion proteins function as potent transcriptional tion factors, produces aberrant transcriptional activators activators (May et al., 1993b; Ohno et al., 1993; that function as dominant oncogenes. In Malignant Bailly et al., 1994; Brown et al., 1995; Lessnick et al., Melanoma of Soft Parts the N-terminal region of 1995; Fujimura et al., 1996) in a manner that is EWS is fused to C-terminal region of the cAMP- dependent on the EWS N-terminal region, hereafter inducible transcription factor ATF1. The EWS/ATF1 referred to as the EWS Activation Domain (EAD). It is fusion protein binds to ATF sites present in cAMP- envisioned that distinct tumors arise via de-regulation responsive promoters via the ATF1 bZIP domain and of dierent genes, depending on the fusion partner for activates transcription constitutively in a manner that is EWS. In cases where it has been examined, agents that dependent on an activation domain (EAD) present in antagonise EWS-fusion proteins also inhibit cellular EWS. To further de®ne the requirements for trans- proliferation (Ouchida et al., 1995; Kovar et al., 1996; activation we have performed mutational analysis of Yi et al., 1997; Tanaka et al., 1997), indicating that EWS/ATF1 in mammalian cells and report several new EWS fusions can play a role in both tumor formation ®ndings. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Atf1 Arid3a Atf2
TFs Datasets Sequence logos of information models showing cofactor motifs Top 500 peaks (FOXA motif) HepG2, Stanford ARID3A (optimal IDR thresholded) Top 5000 peaks (FOXA motif) All 14818 peaks (IRF motif) K562, HMS ATF1 (optimal IDR thresholded) Top 2500 peaks (SP motif) H1-hESC, HAIB ATF2 Protocol: v042211.1 (optimal IDR thresholded) All 1860 peaks (USF motif) GM12878, HAIB Protocol: PCR1x (SPP obtained from Factorbook) All 1670 peaks (SP motif) GM12878, HAIB Protocol: PCR1x (optimal IDR thresholded) 1550 peaks (USF motif), after masking the SP motif All 4803 peaks (SP motif) H1-hESC, HAIB Protocol: v041610.2 (optimal IDR thresholded) 4697 peaks (USF motif), after masking the SP motif ATF3 All 3284 peaks (SP motif) HepG2, HAIB Protocol: v041610.1 (optimal IDR thresholded) 3209 peaks (USF motif), after masking the SP motif All 11098 peaks (SP motif) K562, HAIB, Replicate 1 Protocol: v041610.1 (raw peaks) All 1232 peaks (SP motif) K562, HMS (optimal IDR thresholded) 1210 peaks (USF motif), after masking the SP motif All 4448 peaks (AP1 motif) BAF155/ HeLa-S3, Stanford SMARCC1 (SPP obtained from Factorbook) 1342 peaks (AP1 motif), after using masking BAF170/ HeLa-S3, Stanford SMARCC2 (SPP obtained from Factorbook) All 17809 peaks (IRF motif) GM12878, HAIB Protocol: PCR1x (optimal IDR thresholded) 17321 peaks (RUNX motif), after masking the IRF motif BCL11A All 2513 peaks (SOX2-OCT4 motif) H1-hESC, HAIB Protocol: PCR1x (optimal IDR thresholded) All 7707 peaks (AP1 motif), after masking noise A549, HAIB Protocol: v042211.1 BCL3 Treatment: -
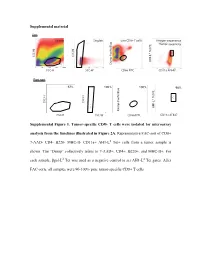
Supplemental Material Supplemental Figure 1. Tumor-Specific CD8+
Supplemental material Sort Scatter Singlets Live CD8+ T cells Antigen-experience Tumor-specifcity c Blue f Tet PE Tet d SSC-H SSC-H AH1-L Dump Paci FSC-H SSC-W CD8α FITC CD11a AF647 Post sort 67% 100%67% 100% 98% c Blue f Tet PE Tet d SSC-H SSC-H AH1-L SSC-Height Dump Paci FSC-H SSC-W CD8α FITC CD11a AF647 SSC-Height Supplemental Figure 1. Tumor-specific CD8+ T cells were isolated for microarray analysis from the timelines illustrated in Figure 2A. Representative FAC-sort of CD8+ 7-AAD- CD4- B220- MHC-II- CD11a+ AH1-Ld Tet+ cells from a tumor sample is shown. The “Dump” collectively refers to 7-AAD+, CD4+, B220+, and MHC-II+. For each sample, βgal-Ld Tet was used as a negative control to set AH1-Ld Tet gates. After FAC-sorts, all samples were 90-100% pure tumor-specific CD8+ T cells. Supplemental Table 1. Transcriptional profiles of published T cell programs compared to tumor-specific TIL T cell program of interest Exhausted (overexpressed) Exhausted (underrexpressed) Memory (overexpressed) Memory (underexpressed) Effector (overexpressed) Effector (underexpressed) Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ Exhausted virus-specific CD8+ cells; also not in memory and Memory virus-specific CD8+ cells; also not in exhausted Effector virus-specific T cells; als not in memory, T cell programs compared for T cells vs. effector vs naïve virus-specific T cells vs. and effector vs naïve virus- CD8+ T cells vs. and exhausted vs naïve relative gene expression CD8+ T cells specific CD8+ T cells virus-specific CD8+ T cells Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ Naïve virus-specific CD8+ cells cells cells cells T cells T cells Reference Wherry EJ et al. -

ABBOTT MOLECULAR ONCOLOGY and GENETICS 2015-2016 Product Catalog
DESCRIPTOR, 9/12, ALL CAPS ABBOTT MOLECULAR ONCOLOGY AND GENETICS 2015-2016 Product Catalog Area for placed imagery Only use imagery that is relevant to the communication CHOOSE TRANSFORMATION See where it will take you at AbbottMolecular.com 2 Please note some products may not be for sale in all markets. Contact your local representative for availability. ASR (Analyte Specific Reagent) Analytical and performance characteristics are not established CE (CE Marked) Conformité Européenne GPR (General Purpose Reagent) For Laboratory use RUO (Research Use Only) Not for use in diagnostic procedures All products manufactured and/or distributed by Abbott Molecular should be used in accordance with the products’ labeled intended use. Products labeled “Research Use Only” should be used for research applications, and are not for use in diagnostic procedures. CEP, LSI, AneuVysion, MultiVysion, PathVysion and Vysis are registered trademarks of Vysis, Inc., AutoVysion, ProbeChek, SpectrumAqua, SpectrumBlue, SpectrumGreen, SpectrumGold, SpectrumOrange, SpectrumRed, TelVysion, ToTelVysion, UroVysion and VP 2000 are trademarks of Abbott Molecular in various jurisdictions. All other trademarks are the property of their respective owners. Please note some products may not be for sale in all markets. Contact your local representative for availability. 3 Abbott Molecular is Transforming Laboratory Partnerships and Productivity—Today and into the Future As a leader in molecular Our commitment to exploring new clinical frontiers is evident in the development and delivery of innovative diagnostics, Abbott is committed systems and assay solutions that aid physicians in the diagnosis of disease, selection of therapies and monitoring to providing solution-oriented of disease. offerings built on FISH and PCR. -

CREB/PKA Sensitive Signalling Pathways Activate and Maintain
1309 HEPATITIS CREB/PKA sensitive signalling pathways activate Gut: first published as 10.1136/gut.2005.065086 on 4 May 2005. Downloaded from and maintain expression levels of the hepatitis B virus pre-S2/S promoter F Tacke, C Liedtke, S Bocklage, M P Manns, C Trautwein ............................................................................................................................... Gut 2005;54:1309–1317. doi: 10.1136/gut.2005.065086 Background and aims: CREB (cAMP response element binding protein) transcription factors are key regulators of homeostatic functions in the liver, and CRE binding is increased in hepatic inflammation. See end of article for During chronic hepatitis B virus (HBV) infection, mutations or deletions in the pre-S region are frequently authors’ affiliations observed. These mutations can affect the pre-S2/S promoter controlling HBV envelope protein expression ....................... (hepatitis B surface antigen (HBsAg)) and have been associated with worsened clinical outcome. We Correspondence to: aimed to test if CREB activation impacts on HBsAg expression. Dr C Trautwein, Hannover Methods: The effect of the CREB inducer protein kinase A (PKA) was tested by coexpression with HBV wild- Medical School, Department of type vector in vitro. Luciferase reporter gene constructs were cloned to identify novel regulatory regions for Gastroenterology, the HBV pre-S2/S promoter. Electrophoretic mobility shift assay (EMSA) gelshift and supershift Hepatology, and experiments were conducted to confirm DNA transcription factor binding. Endocrinology, Carl- Neuberg-Strasse 1, Results: Coexpression of HBV and PKA resulted in HBV-S mRNA induction and enhanced small envelope D-30625 Hannover, protein expression. We identified a CREB binding motif in the transcribed part of the pre-S2 region, Germany; contributing to basal S promoter activity via binding of activating transcription factor 2 (ATF2). -

Xo PANEL DNA GENE LIST
xO PANEL DNA GENE LIST ~1700 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes (at 400 -500x average coverage on tumor) Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 -

An Evaluation of Cancer Subtypes and Glioma Stem Cell Characterisation Unifying Tumour Transcriptomic Features with Cell Line Expression and Chromatin Accessibility
An evaluation of cancer subtypes and glioma stem cell characterisation Unifying tumour transcriptomic features with cell line expression and chromatin accessibility Ewan Roderick Johnstone EMBL-EBI, Darwin College University of Cambridge This dissertation is submitted for the degree of Doctor of Philosophy Darwin College December 2016 Dedicated to Klaudyna. Declaration • I hereby declare that except where specific reference is made to the work of others, the contents of this dissertation are original and have not been submitted in whole or in part for consideration for any other degree or qualification in this, or any other university. • This dissertation is my own work and contains nothing which is the outcome of work done in collaboration with others, except as specified in the text and Acknowledge- ments. • This dissertation is typeset in LATEX using one-and-a-half spacing, contains fewer than 60,000 words including appendices, footnotes, tables and equations and has fewer than 150 figures. Ewan Roderick Johnstone December 2016 Acknowledgements This work was funded by the Biotechnology and Biological Sciences Research Council (BBSRC, Ref:1112564) and supported by the European Molecular Biology Laboratory (EMBL) and its outstation, the European Bioinformatics Institute (EBI). I have many people to thank for assistance in preparing this thesis. First and foremost I must thank my supervisor, Paul Bertone for his support and willingness to take me on as a student. My thanks are also extended to present and past members of the Bertone group, particularly Pär Engström and Remco Loos who have provided a great deal of guidance over the course of my studentship. -

Subtype Heterogeneity and Epigenetic Convergence in Neuroendocrine Prostate Cancer
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.13.291328; this version posted September 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Subtype Heterogeneity and Epigenetic Convergence in Neuroendocrine Prostate Cancer Paloma Cejas1,2,3, Yingtian Xie1,2, Alba Font-Tello1,2, Klothilda Lim1,2, Sudeepa Syamala1,2, Xintao Qiu1,2, Alok K. Tewari1,2,4, Neel Shah1,2, Holly M Nguyen5, Radhika A. Patel6, Lisha Brown5, Ilsa Coleman6, Wenzel M. Hackeng7, Lodewijk Brosens7, Koen M.A. Dreijerink8, Leigh Ellis4,9, Sarah Abou Alaiwi1, Ji-Heui Seo1, Mark Pomerantz1, Alessandra Dall'Agnese10, Jett Crowdis1,4, Eliezer M. Van Allen1,4, Joaquim Bellmunt11, Colm Morrisey5, Peter S. Nelson6, James DeCaprio1, Anna Farago12, Nicholas Dyson12, Benjamin Drapkin13,14,15, X. Shirley Liu2,16, Matthew Freedman1,2, Michael C. Haffner6,17,18, Eva Corey5, Myles Brown1,2* and Henry W. Long1,2* 1. Department of Medical Oncology, Dana-Farber Cancer Institute, Brigham and Women’s Hospital, and Harvard Medical School, Boston, Massachusetts, USA. 2. Center for Functional Cancer Epigenetics, Dana-Farber Cancer Institute, Boston, Massachusetts, USA. 3. Translational Oncology Laboratory, Hospital La Paz Institute for Health Research (IdiPAZ) and CIBERONC, La Paz University Hospital, Madrid, Spain. 4. Broad Institute of MIT and Harvard, Cambridge, MA, USA 5. Department of Urology, University of Washington, Seattle, WA, USA 6. Divisions of Human Biology and Clinical Research, Fred Hutchinson Cancer Research Center, Seattle, WA, USA 7.