Occupancy Maps of 208 Chromatin-Associated Proteins In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -
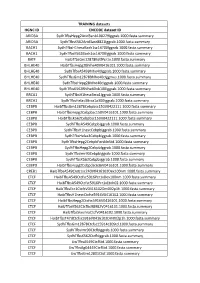
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Akt1 and Dciz1 Promote Cell Survival from Apoptotic Caspase Activation
ARTICLE https://doi.org/10.1038/s41467-020-19068-2 OPEN Akt1 and dCIZ1 promote cell survival from apoptotic caspase activation during regeneration and oncogenic overgrowth ✉ ✉ Gongping Sun1,2 , Xun Austin Ding2, Yewubdar Argaw2, Xiaoran Guo 2 & Denise J. Montell 2 Apoptosis is an ancient and evolutionarily conserved cell suicide program. During apoptosis, executioner caspase enzyme activation has been considered a point of no return. However, 1234567890():,; emerging evidence suggests that some cells can survive caspase activation following expo- sure to apoptosis-inducing stresses, raising questions as to the physiological significance and underlying molecular mechanisms of this unexpected phenomenon. Here, we show that, following severe tissue injury, Drosophila wing disc cells that survive executioner caspase activation contribute to tissue regeneration. Through RNAi screening, we identify akt1 and a previously uncharacterized Drosophila gene CG8108, which is homologous to the human gene CIZ1, as essential for survival from the executioner caspase activation. We also show that cells expressing activated oncogenes experience apoptotic caspase activation, and that Akt1 and dCIZ1 are required for their survival and overgrowth. Thus, survival following executioner caspase activation is a normal tissue repair mechanism usurped to promote oncogene-driven overgrowth. 1 The Key Laboratory of Experimental Teratology, Ministry of Education and Department of Anatomy and Histoembryology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China. 2 Molecular, Cellular, and Developmental Biology Department, University of ✉ California, Santa Barbara, CA 93106, USA. email: [email protected]; [email protected] NATURE COMMUNICATIONS | (2020) 11:5726 | https://doi.org/10.1038/s41467-020-19068-2 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-19068-2 recise control of cell death and survival is critical for tissue we took advantage of the CasExpress biosensor11. -

A Genome-Wide Analysis of the ERF Gene Family in Sorghum
A genome-wide analysis of the ERF gene family in sorghum H.W. Yan, L. Hong, Y.Q. Zhou, H.Y. Jiang, S.W. Zhu, J. Fan and B.J. Cheng Key Laboratory of Crop Biology of Anhui Province, Anhui Agricultural University, Hefei, China Corresponding author: B.J. Cheng E-mail: [email protected] Genet. Mol. Res. 12 (2): 2038-2055 (2013) Received November 9, 2012 Accepted March 8, 2013 Published May 13, 2013 DOI http://dx.doi.org/10.4238/2013.May.13.1 ABSTRACT. The ethylene response factor (ERF) family are members of the APETALA2 (AP2)/ERF transcription factor superfamily; they are known to play an important role in plant adaptation to biotic and abiotic stress. ERF genes have been studied in Arabidopsis, rice, grape, and maize; however, there are few reports of ERF genes in sorghum. We identified 105 sorghum ERF (SbERF) genes, which were categorized into 12 groups (A-1 to A-6 and B-1 to B-6) based on their sequence similarity, and this new method of classification for ERF genes was then further characterized. A comprehensive bioinformatic analysis of SbERF genes was performed using a sorghum genomic database, to analyze the phylogeny of SbERF genes, identify other conserved motifs apart from the AP2/ERF domain, map SbERF genes to the 10 sorghum chromosomes, and determine the tissue-specific expression patterns of SbERF genes. Gene clustering indicates that SbERF genes were generated by tandem duplications. Comparison of SbERF genes with maize ERF homologs suggests lateral gene transfer between monocot species. These results can contribute to our understanting of Genetics and Molecular Research 12 (2): 2038-2055 (2013) ©FUNPEC-RP www.funpecrp.com.br Analysis of the ERF gene family in sorghum 2039 the evolution of the ERF gene family. -

Maintenance of Epigenetic Landscape Requires CIZ1 and Is Corrupted in Differentiated fibroblasts in Long-Term Culture
ARTICLE https://doi.org/10.1038/s41467-018-08072-2 OPEN Maintenance of epigenetic landscape requires CIZ1 and is corrupted in differentiated fibroblasts in long-term culture Emma R. Stewart1, Robert M.L. Turner1, Katherine Newling 2, Rebeca Ridings-Figueroa1,3, Victoria Scott1, Peter D. Ashton2, Justin F.X. Ainscough 1 & Dawn Coverley1 1234567890():,; The inactive X chromosome (Xi) serves as a model for establishment and maintenance of repressed chromatin and the function of polycomb repressive complexes (PRC1/2). Here we show that Xi transiently relocates from the nuclear periphery towards the interior during its replication, in a process dependent on CIZ1. Compromised relocation of Xi in CIZ1-null primary mouse embryonic fibroblasts is accompanied by loss of PRC-mediated H2AK119Ub1 and H3K27me3, increased solubility of PRC2 catalytic subunit EZH2, and genome-wide deregulation of polycomb-regulated genes. Xi position in S phase is also corrupted in cells adapted to long-term culture (WT or CIZ1-null), and also accompanied by specific changes in EZH2 and its targets. The data are consistent with the idea that chromatin relocation during S phase contributes to maintenance of epigenetic landscape in primary cells, and that elevated soluble EZH2 is part of an error-prone mechanism by which modifying enzyme meets template when chromatin relocation is compromised. 1 Department of Biology, University of York, York YO10 5DD, UK. 2 York Bioscience Technology Facility, University of York, York YO10 5DD, UK. 3Present address: Department of Genetics, University of Cambridge, Cambridge CB2 3EH, UK. Correspondence and requests for materials should be addressed to D.C. (email: [email protected]) NATURE COMMUNICATIONS | (2019) 10:460 | https://doi.org/10.1038/s41467-018-08072-2 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-018-08072-2 he inactive X chromosome (Xi) is a discrete unit of attachment at Xi is by association with RNA5, most likely Xist6,17. -

The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc Oncogenesis
The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis By Yuting Sun This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy at the University of New South Wales Children’s Cancer Institute Australia for Medical Research School of Women’s and Children’s Health, Faculty of Medicine University of New South Wales Australia August 2014 PLEASE TYPE THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Sun First name: Yuting Other name/s: Abbreviation for degree as given in the University calendar: PhD School : School of·Women's and Children's Health Faculty: Faculty of Medicine Title: The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis. Abstract 350 words maximum: (PLEASE TYPE) N-Myc Induces neuroblastoma by regulating the expression of target genes and proteins, and N-Myc protein is degraded by Fbxw7 and NEDD4 and stabilized by Aurora A. The class lla histone deacetylase HDAC5 suppresses gene transcription, and blocks myoblast and leukaemia cell differentiation. While histone H3 lysine 4 (H3K4) trimethylation at target gene promoters is a pre-requisite for Myc· induced transcriptional activation, WDRS, as a histone H3K4 methyltransferase presenter, is required for H3K4 methylation and transcriptional activation mediated by a histone H3K4 methyltransferase complex. Here, I investigated the roles of HDAC5 and WDR5 in N-Myc overexpressing neuroblastoma. I have found that N-Myc upregulates HDAC5 protein expression, and that HDAC5 represses NEDD4 gene expression, increases Aurora A gene expression and consequently upregulates N-Myc protein expression in neuroblastoma cells. -

(RRP1B) Modulates Metastasis Through Regulation of Histone Methylation
Published OnlineFirst August 4, 2014; DOI: 10.1158/1541-7786.MCR-14-0167 Molecular Cancer Oncogenes and Tumor Suppressors Research Metastasis-Associated Protein Ribosomal RNA Processing 1 Homolog B (RRP1B) Modulates Metastasis through Regulation of Histone Methylation Minnkyong Lee1, Amy M. Dworkin1, Jens Lichtenberg1, Shashank J. Patel1, Niraj S. Trivedi2, Derek Gildea2, David M. Bodine1, and Nigel P.S. Crawford1 Abstract Overexpression of ribosomal RNA processing 1 homolog B (RRP1B) induces a transcriptional profile that accurately predicts patient outcome in breast cancer. However, the mechanism by which RRP1B modulates transcription is unclear. Here, the chromatin-binding properties of RRP1B were examined to define how it regulates metastasis-associated transcription. To identify genome-wide RRP1B-binding sites, high-throughput ChIP-seq was performed in the human breast cancer cell line MDA-MB-231 and HeLa cells using antibodies against endogenous RRP1B. Global changes in repressive marks such as histone H3 lysine 9 trimethylation (H3K9me3) were also examined by ChIP-seq. Analysis of these samples identified 339 binding regions in MDA- MB-231 cells and 689 RRP1B-binding regions in HeLa cells. Among these, 136 regions were common to both cell lines. Gene expression analyses of these RRP1B-binding regions revealed that transcriptional repression is the primary result of RRP1B binding to chromatin. ChIP-reChIP assays demonstrated that RRP1B co-occupies loci with decreased gene expression with the heterochromatin-associated proteins, tripartite motif-containing protein 28 (TRIM28/KAP1), and heterochromatin protein 1-a (CBX5/HP1a). RRP1B occupancy at these loci was also associated with higher H3K9me3 levels, indicative of heterochromatinization mediated by the TRIM28/HP1a complex. -

ARID5B As a Critical Downstream Target of the TAL1 Complex That Activates the Oncogenic Transcriptional Program and Promotes T-Cell Leukemogenesis
Downloaded from genesdev.cshlp.org on October 10, 2021 - Published by Cold Spring Harbor Laboratory Press ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis Wei Zhong Leong,1 Shi Hao Tan,1 Phuong Cao Thi Ngoc,1 Stella Amanda,1 Alice Wei Yee Yam,1 Wei-Siang Liau,1 Zhiyuan Gong,2 Lee N. Lawton,1 Daniel G. Tenen,1,3,4 and Takaomi Sanda1,4 1Cancer Science Institute of Singapore, National University of Singapore, 117599 Singapore; 2Department of Biological Sciences, National University of Singapore, 117543 Singapore; 3Harvard Medical School, Boston, Massachusetts 02215, USA; 4Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, 117599 Singapore The oncogenic transcription factor TAL1/SCL induces an aberrant transcriptional program in T-cell acute lym- phoblastic leukemia (T-ALL) cells. However, the critical factors that are directly activated by TAL1 and contribute to T-ALL pathogenesis are largely unknown. Here, we identified AT-rich interactive domain 5B (ARID5B) as a col- laborating oncogenic factor involved in the transcriptional program in T-ALL. ARID5B expression is down-regulated at the double-negative 2–4 stages in normal thymocytes, while it is induced by the TAL1 complex in human T-ALL cells. The enhancer located 135 kb upstream of the ARID5B gene locus is activated under a superenhancer in T-ALL cells but not in normal T cells. Notably, ARID5B-bound regions are associated predominantly with active tran- scription. ARID5B and TAL1 frequently co-occupy target genes and coordinately control their expression. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

The Alternative Role of DNA Methylation in Splicing Regulation
TIGS-1191; No. of Pages 7 Review The alternative role of DNA methylation in splicing regulation Galit Lev Maor, Ahuvi Yearim, and Gil Ast Department of Human Molecular Genetics and Biochemistry, Sackler Medical School, Tel Aviv University, Tel Aviv, Israel Although DNA methylation was originally thought to only Alternative splicing is an evolutionarily conserved mech- affect transcription, emerging evidence shows that it also anism that increases transcriptome and proteome diversity regulates alternative splicing. Exons, and especially splice by allowing the generation of multiple mRNA products from sites, have higher levels of DNA methylation than flanking a single gene [10]. More than 90% of human genes were introns, and the splicing of about 22% of alternative exons shown to undergo alternative splicing [11,12]. Furthermore, is regulated by DNA methylation. Two different mecha- the average number of spliced isoforms per gene is higher in nisms convey DNA methylation information into the vertebrates [13], implying that the prevalence of alternative regulation of alternative splicing. The first involves mod- splicing in these organisms is important for their greater ulation of the elongation rate of RNA polymerase II (Pol II) complexity. The splicing reaction is regulated by various by CCCTC-binding factor (CTCF) and methyl-CpG binding activating and repressing elements such as cis-acting se- protein 2 (MeCP2); the second involves the formation of a quence signals and RNA-binding proteins [13–15]. Its regu- protein bridge by heterochromatin protein 1 (HP1) that lation is essential for providing cells and tissues their recruits splicing factors onto transcribed alternative specific features, and for their response to environmental exons. -

A Flexible Microfluidic System for Single-Cell Transcriptome Profiling
www.nature.com/scientificreports OPEN A fexible microfuidic system for single‑cell transcriptome profling elucidates phased transcriptional regulators of cell cycle Karen Davey1,7, Daniel Wong2,7, Filip Konopacki2, Eugene Kwa1, Tony Ly3, Heike Fiegler2 & Christopher R. Sibley 1,4,5,6* Single cell transcriptome profling has emerged as a breakthrough technology for the high‑resolution understanding of complex cellular systems. Here we report a fexible, cost‑efective and user‑ friendly droplet‑based microfuidics system, called the Nadia Instrument, that can allow 3′ mRNA capture of ~ 50,000 single cells or individual nuclei in a single run. The precise pressure‑based system demonstrates highly reproducible droplet size, low doublet rates and high mRNA capture efciencies that compare favorably in the feld. Moreover, when combined with the Nadia Innovate, the system can be transformed into an adaptable setup that enables use of diferent bufers and barcoded bead confgurations to facilitate diverse applications. Finally, by 3′ mRNA profling asynchronous human and mouse cells at diferent phases of the cell cycle, we demonstrate the system’s ability to readily distinguish distinct cell populations and infer underlying transcriptional regulatory networks. Notably this provided supportive evidence for multiple transcription factors that had little or no known link to the cell cycle (e.g. DRAP1, ZKSCAN1 and CEBPZ). In summary, the Nadia platform represents a promising and fexible technology for future transcriptomic studies, and other related applications, at cell resolution. Single cell transcriptome profling has recently emerged as a breakthrough technology for understanding how cellular heterogeneity contributes to complex biological systems. Indeed, cultured cells, microorganisms, biopsies, blood and other tissues can be rapidly profled for quantifcation of gene expression at cell resolution.