Improved Processing Isoolefin Polymers Verarbeitung Von Isoolefin-Polymeren Transformation De Polymeres D'iso-Olefine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry Lesson 10 Reactions of Organic Molecules Combusition Reactions Substitution Reactions
Organic Chemistry Lesson 10 Reactions of Organic Molecules Combusition Reactions Substitution Reactions Physical Sciences Grade 12 1 Instructions Lesson 10 (16 April 2020) 1. Read ALL the slides in this document. 2. Homework: Just understand the theory for now. • If you have any questions, post them onto your WhatsApp group and your teacher will attend to them when (s)he has a chance. • Additional Resources: • Summary on pg.132 of your textbook. Physical Sciences Grade 12 2 Types of Reactions of Organic Compounds 1. Esterification: formation of an ester from an alcohol and a carboxylic acid. 2. Combustion Reactions (aka Oxidation): organic molecule + oxygen. 3. Substitution Reactions: one or more atoms are replaced by other atoms. (i) alkanes to alkyl halides; (ii) alcohols to alkyl halides; (iii) alkyl halides to alcohols 4. Addition Reactions: one or more atoms are added. (i) hydrogenation; (ii) halogenation; (iii) hydrohalogenation; (iv) hydration 5. Elimination Reactions: one or more atoms are removed. (i) dehydrohalogenation; (ii) dehydration; (iii) cracking of alkanes Physical Sciences Grade 12 3 1. Esterification • Already discussed (refer to earlier notes). Physical Sciences Grade 12 4 2. Combustion Reactions, i.e. Oxidation • A combustion reaction is the reaction of an organic molecule with oxygen. • Please note: “combustion” does NOT mean “explosion”. All combustion reactions release energy but most do so WITHOUT any explosion. • Alkanes – in the form of fossil fuels – are currently the main source of energy worldwide as their combustion is highly exothermic. • Examples: Burning wood for a braai. Propane combusting in a gas stove. Petrol combusting in a car engine. Physical Sciences Grade 12 5 2. -

Activity 33. Hydrohalogenation & Hydration of Alkynes
Chem 201, Fall 2010 Activity 33, M Nov 22 Activity 33. Hydrohalogenation & Hydration of Alkynes Alkynes undergo many of the same addition reactions that alkenes do, including additions that require carbocation intermediates. Different energy and geometry changes may be required for additions to an alkyne and an alkene so some surprising outcomes may occur with alkynes. Model 1. Addition of HX Hydrogen halides (HCl, HBr, HI) add to alkynes to make vinyl halides . A carbocation mechanism is followed when the reaction is performed in the dark in a peroxide-free solvent. This addition obeys Markovnikov’s rule. A free-radical-chain mechanism is followed when HBr addition is initiated by organic peroxides. This addition gives an anti-Markonikov product. Critical Thinking Questions 1. Draw the carbocations indicated by the two bouncy curved arrows (below). Based on the observation that the carbocation mechanism yields a Markovnikov product, circle the more stable carbocation. 2. (CI) Draw the free radicals indicated by the two competing pathways (below). Based on the observation that the free-radical mechanism yields an anti-Markovnikov product, circle the more stable free radical. 3. The cations and radicals in CTQ #1 and #2 are called vinyl cations and vinyl radicals , respectively. Based on the regioselectivities reported in Model 1, what effect do alkyl substituents at the charged/radical carbon have on the energies of these species? Is this consistent with, or opposed to, their effect on R 3C+ and R 3C• species? 4. The geometry of a vinyl cation can be anticipated using VSEPR. Count electron domains in the cation below and predict all of the HCC bond angles. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

UNIT-III: Mechanistic and Sterochemical Aspects of Addition Reaction Involving Electrophiles & Nucleophiles
MCH-204:ORGANIC CHEMISTRY UNIT-III: Mechanistic and sterochemical aspects of addition reaction Involving electrophiles & nucleophiles Prof.Anand Halve S.O.S in Chemistry Jiwaji University Gwalior ELECTROPHILIC ADDITION REACTIONS markovnikov’ additions markovnikov’s rule Addition of hydrogen to an unsymmetrical olefin occurs at those carbon atoms with maximum number of hydrogen atoms. (i.e., the carbon with least substitution). Electronegative group goes to more substituted carbon atom. Such an addition leads to a stabler carbocation. Such a reaction may lead to constitutional isomers but actually one of the products is formed as major product. X X Formed H HX X=F, Cl. X Olefin Not formed origin … X HX X X=F, Cl . carbocation is more stablised in T.S. Stereo specific product H X X carbocation is not enough stablilised in transition state Olefin Consider two possible sites for hydrogen addition (i) terminal or (ii) internal (substituted carbon). The addition of hydrogen at the terminal carbon leads to better stabilization of carbocation, the chances of stabilization increases with increase in conjugation with olefin. The terminal carbocation require higher activation energy which is not a favorable condition, leading to slower reaction rate. However, the generation of non terminal carbocation is assisted by hyperconjugative stabilization leading to a lower activation energy. Alkenes-some facts Due to trigonal planar geometry of olefin carbon atoms the addition can occur on the same side (syn periplanar) or on opposite sides (anti periplanar). Alkenes are generally nucleophilic. The C=C double bond provides a higher energy HOMO (highest occupied molecular orbitals). Electron donating groups increase the rate for electrophilic attack as they assist in carbocation and positive charge stabilization in the TS. -
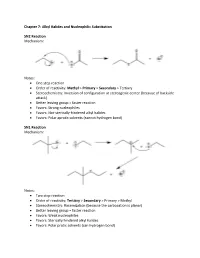
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Reaction-Of-Alkenes.Pdf
8 Reactions of Alkenes Goals for Chapter 8 1 Explain why electrophilic additions are among the most common reactions of alkenes. femoral component 2 Predict the products of the reactions of alkenes, including the orientation of the reaction (regio- polyethylene bearing chemistry) and the stereochemistry. tibial plate 3 Propose mechanisms to explain the observed products of alkene reactions. 4 Use retrosynthetic analysis to solve multistep synthesis problems with alkenes as reagents, intermediates, or products. ◀ Polyethylene provides a self-lubricating surface for movement of the metal parts of an artificial knee replacement. The highly cross-linked polyethylene used in these implants is fabricated to be exceptionally tough: It wears only about 0.1 mm per year. Polyethylene is compatible with the human body, and in most cases, it does not cause a foreign body reaction even after years of constant movement in the joint. The alkene double bond is a gateway functional group. Alkene reactions lead to many other functional groups that lay the foundation for the rest of your study of organic chemistry. You can convert alkenes to alkyl halides, epoxides, alcohols, aldehydes, ketones, carboxylic acids, and other functional groups. The reactions of alkenes arise from the reactivity of their carbon–carbon double bonds. Organic chemists enjoy the challenge of taking a simple carbon–carbon double bond and manipulating it in all possible ways to produce other compounds, often mimicking biological reactions that occur in cells. This chapter covers the most common alkene reactions, including their mechanisms, reactivity, orientation, and stereochemistry. Most reactions of alkenes involve addition of atoms or groups across the double bond, with one atom or group adding to each end. -

Hydrohalogenation
Hydrohalogenation A hydrohalogenation reaction is the electrophilic addition of hydrohalic acids like hydrogen chloride or hydrogen bromide to alkenes to yield the corresponding haloalkanes.[1][2][3] If the two carbon atoms at the double bond are linked to a different number of hydrogen atoms, the halogen is found preferentially at the carbon with fewer hydrogen substituents, an observation known as Markovnikov's rule. This is due to the abstraction of a hydrogen atom by the alkene from the acid (HX) to form the most stable carbocation (relative stability: 3°>2°>1°>methyl), as well as generating a halogen anion. A simple example of a hydrochlorination is that of indene with hydrogen chloride gas (no solvent):[4] Alkynes also undergo hydrohalogenation reactions. Depending on the exact substrate, alkyne hydrohalogenation can proceed though a concerted protonation/nucleophilic attack (AdE3) or stepwise by first - protonating the alkyne to form a vinyl cation, followed by attack of HX/X to give the product (AdE2) (see electrophile for arrow pushing).[5] As in the case of alkenes, the regioselectivity is determined by the relative ability of the carbon atoms to stabilize positive charge (either a partial charge in the case of a concerted transition state or a full formal charge for a discrete vinyl cation). Depending on reaction conditions, the main product could be this initially formed alkenyl halide, or the product of twice hydrohalogenation to form a dihaloalkane. In most cases, the main regioisomer formed is the gem-dihaloalkane.[6] This regioselectivity is rationalized by the resonance stabilization of a neighboring carbocation by a lone pair on the initially installed halogen. -

United States Patent Office Patented Dec
2,774,785 United States Patent Office Patented Dec. 18, 1956 1. the acids that are suitable in the process of the present 2,774,785 invention: acrylic acid, maleic acid, fumaric acid. The following are representative of the esters that are ADDITION OF HYDROGEN CHLORIDE OR HY. Suitable in the process of the present invention: methyl DROGEN BROMIDE TO MALEIC, FUMARIC, OR acrylate, ethyl acrylate, butyl acrylate, isobutyl acrylate, ACRYLIC ACDS OR THER ESTERS beta methoxyethyl acrylate, benzyl acrylate, phenethyl Vernon P. Wystrach, Stamford. Conn., assignor to Ameri acrylate, 2-thionyl acrylate, alpha-chloro acrylic acid, can Cyanamid Company, New York, N.Y., a corpora ethyl alpha-chloro acrylate, mono- and dimethyl fumarate, tion of Maine mono- and dimethyl maleate, mono- and diethyl fumarate, No Drawing. Application February 23, 1954, 0 mono- and diethyl maleate, mono- and dibutyl fumarate, mono- and dibutyl maleate, and other maleic and fumaric Serial No. 412,067 acid derivatives having of unsaturation. 8 Claims. (C. 260-485) The hydrohalogenation process is carried out with vary ing degrees of ease depending on the alpha-beta un This invention relates to hydrohalogenation and more 5 Saturated compound. A diester hydrohalogenates easiest; particularly to a process for adding a hydrogen halide to a monoester of a dibasic compound hydrohalogenates an alpha-beta unsaturated compound selected from the next easiest; an ester of a monobasic acid hydrohalo group consisting of acids and esters. genates with a little more difficulty; and the acids hydro Hydrogen halides under normal conditions add to alpha halogenate with the greatest difficulty. beta unsaturated compounds only with difficulty. -

An Investigation Into Synthetic Routes to Optically Active Trityl Systems
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 1-1-1964 An investigation into synthetic routes to optically active trityl systems. Joseph M. Prokipcak University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Prokipcak, Joseph M., "An investigation into synthetic routes to optically active trityl systems." (1964). Electronic Theses and Dissertations. 6037. https://scholar.uwindsor.ca/etd/6037 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. AN INVESTIGATION INTO SYNTHETIC ROUTES TO I OPTICALLY ACTIVE TRITYL SYSTEMS BY JOSEPH M. PROKIPCAK A Thesis Submitted to the Faculty of Graduate Studies through the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the University of Windsor Windsor, Ontario 1964 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. -

The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-Closo-Hexaborate Dianion
molecules Article The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion Igor E. Golub * , Oleg A. Filippov , Natalia V. Belkova , Lina M. Epstein and Elena S. Shubina * A. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences (INEOS RAS), 28, Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (N.V.B.); [email protected] (L.M.E.) * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) − Abstract: The mechanism of the consecutive halogenation of the tetrahydroborate anion [BH4] by 2− hydrogen halides (HX, X = F, Cl, Br) and hexahydro-closo-hexaborate dianion [B6H6] by HCl via electrophile-induced nucleophilic substitution (EINS) was established by ab initio DFT calculations [M06/6-311++G(d,p) and wB97XD/6-311++G(d,p)] in acetonitrile (MeCN), taking into account non- specific solvent effects (SMD model). Successive substitution of H− by X− resulted in increased electron deficiency of borohydrides and changes in the character of boron atoms from nucleophilic to highly electrophilic. This, in turn, increased the tendency of the B–H bond to transfer a proton rather than a hydride ion. Thus, the regularities established suggested that it should be possible to carry out halogenation more selectively with the targeted synthesis of halogen derivatives with a low degree of substitution, by stabilization of H2 complex, or by carrying out a nucleophilic substitution of B–H bonds activated by interaction with Lewis acids (BL3). Citation: Golub, I.E.; Filippov, O.A.; Keywords: borohydride; polyhedral closo-borane; proton transfer; Lewis acidity; hydride donor Belkova, N.V.; Epstein, L.M.; Shubina, ability; DFT calculations E.S. -

Level 2 Organic Chemistry
No Brain Too Small CHEMISTRY AS 91165 LEVEL 2 ORGANIC CHEMISTRY Place in a molecule where a reaction takes place. Compounds with the Naming (IUPAC nomenclature) same functional group – homologous series - have similar properties. First the functional groups… alkane alkene alkyne haloalkane -ane -ene -yne bromo, chloro etc H H H H C H H H H H C H H H H H C C C C H H C Br C C H C C C C H H H H H H H C H C C H H H C H H H H H H H alcohol carboxylic acid amine -anol -anoic acid amino C-C C=C C≡C H H H H O O R-X H H H H H H H C C N H C C C C C H H C C C R-OH H H H H H H H R-COOH H C H H H O H H R-NH2 No Brain Too Small CHEMISTRY AS 91165 What functional groups can you identify here? No Brain Too Small CHEMISTRY AS 91165 N a m I n g No. of carbons Alkane name Prefix Side chain 1 methane meth- methyl- 2 ethane eth- ethyl- 3 propane prop- propyl- 4 butane but- butyl- 5 pentane pent- 6 hexane hex- 7 heptane hept- 8 octane oct- B u t a n – 2 – o l or 2-butanol H H H H H C C C C H H H H O H • Find longest carbon chain – base name on the parent alkane • Number the C atoms in the chain to indicate position of any side chains or functional groups (number from end to give the lowest numbers) • Give names and positions of functional groups eg 3-bromo, and 4-chloro (alphabetically) • Where there’s more than one functional group there is priority order for naming - ol > halo- -anoic acid > halo -anoic acid > hydroxy (-OH) H H H H O H C C C C C H H H O O H H __ - hydroxy ______anoic acid No Brain Too Small CHEMISTRY AS 91165 Name me… Draw me… -

United States Patent (19) 11 Patent Number: 5,071,913 Powers Et Al
United States Patent (19) 11 Patent Number: 5,071,913 Powers et al. 45) Date of Patent: Dec. 10, 1991 54 RUBBERY ISOOLEFIN POLYMERS duction, Chemistry of Natural Rubber, pp. 378-412, EXHIBITING IMPROVED 1960. PROCESSABILITY L. M. White et al., Industrial and Engineering Chemis 75 Inventors: Kenneth W. Powers, Berkeley try, Aug. 1948, "Gel as a Definitive Property in GR-S Heights; Hsien-Chang Wang, Edison; Technology", vol. 37, No. 8, pp. 770-775. Debra C. Handy, Basking Ridge; R. A. Crawford, G. J. Tiger, Industrial and Engineering Janes V. Fusco, Red Bank, all of N.J. Chemistry, vol. 41, No. 3, "Improved-Processing GR-S by Variations in Compounding", Mar. 1949, pp. 73) Assignee: Exxon Chemical Patents Inc., 592-596. Linden, N.J. D. L. Schoene et al., Industrial and Engineering Chem (21) Appl. No.: 131,938 istry, "Development of a Better Processing GR-S", (22 Filed: Dec. 11, 1987 Dec. 1946, vol. 38, No. 12, pp. 1246-1249. 51) Int. Cl. ........................ C08L9/06; CO8L 47/OO; Primary Examiner-Fred Teskin CO8L 51/04; CO8L 23/22 Attorney, Agent, or Firm-H. L. Cohen (52) U.S. Cl. ........................................ 525/87; 525/86; 57 ABSTRACT 525/98; 525/99: 525/191; 525/192: 525/193; A substantially gel free C4 to C7 isoolefin homopolymer 525/313; 525/314: 525/315; 525/319; 525/332.3; 525/333.4; 525/918; 526/339; rubber, butyl copolymer rubber, halogenated butyl 526/348.7; 52/525 rubber (e.g., chlorinated or brominated), or mixtures 58) Field of Search ............ 526/335, 339, 340, 348.7; thereof, comprising a molecular weight distribution 525/98, 99, 86, 87, 193, 332.3, 191, 192,314, such that the ratio of the moments of said molecular 315, 313, 319, 333.4 weight distribution, Mz/Mw, is equal to or exceeds 2.0, and that portion of said molecular weight distribution (56) References Cited which is equal to and greater than 4 times the peak U.S.