ADDITION REACTIONS • Key Words: Addition to Alkenes and Alkynes, Markovnikov’S Rule, Electrophilic and Nucleophilic Additions Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Organic Chemistry Lesson 10 Reactions of Organic Molecules Combusition Reactions Substitution Reactions
Organic Chemistry Lesson 10 Reactions of Organic Molecules Combusition Reactions Substitution Reactions Physical Sciences Grade 12 1 Instructions Lesson 10 (16 April 2020) 1. Read ALL the slides in this document. 2. Homework: Just understand the theory for now. • If you have any questions, post them onto your WhatsApp group and your teacher will attend to them when (s)he has a chance. • Additional Resources: • Summary on pg.132 of your textbook. Physical Sciences Grade 12 2 Types of Reactions of Organic Compounds 1. Esterification: formation of an ester from an alcohol and a carboxylic acid. 2. Combustion Reactions (aka Oxidation): organic molecule + oxygen. 3. Substitution Reactions: one or more atoms are replaced by other atoms. (i) alkanes to alkyl halides; (ii) alcohols to alkyl halides; (iii) alkyl halides to alcohols 4. Addition Reactions: one or more atoms are added. (i) hydrogenation; (ii) halogenation; (iii) hydrohalogenation; (iv) hydration 5. Elimination Reactions: one or more atoms are removed. (i) dehydrohalogenation; (ii) dehydration; (iii) cracking of alkanes Physical Sciences Grade 12 3 1. Esterification • Already discussed (refer to earlier notes). Physical Sciences Grade 12 4 2. Combustion Reactions, i.e. Oxidation • A combustion reaction is the reaction of an organic molecule with oxygen. • Please note: “combustion” does NOT mean “explosion”. All combustion reactions release energy but most do so WITHOUT any explosion. • Alkanes – in the form of fossil fuels – are currently the main source of energy worldwide as their combustion is highly exothermic. • Examples: Burning wood for a braai. Propane combusting in a gas stove. Petrol combusting in a car engine. Physical Sciences Grade 12 5 2. -

Activity 33. Hydrohalogenation & Hydration of Alkynes
Chem 201, Fall 2010 Activity 33, M Nov 22 Activity 33. Hydrohalogenation & Hydration of Alkynes Alkynes undergo many of the same addition reactions that alkenes do, including additions that require carbocation intermediates. Different energy and geometry changes may be required for additions to an alkyne and an alkene so some surprising outcomes may occur with alkynes. Model 1. Addition of HX Hydrogen halides (HCl, HBr, HI) add to alkynes to make vinyl halides . A carbocation mechanism is followed when the reaction is performed in the dark in a peroxide-free solvent. This addition obeys Markovnikov’s rule. A free-radical-chain mechanism is followed when HBr addition is initiated by organic peroxides. This addition gives an anti-Markonikov product. Critical Thinking Questions 1. Draw the carbocations indicated by the two bouncy curved arrows (below). Based on the observation that the carbocation mechanism yields a Markovnikov product, circle the more stable carbocation. 2. (CI) Draw the free radicals indicated by the two competing pathways (below). Based on the observation that the free-radical mechanism yields an anti-Markovnikov product, circle the more stable free radical. 3. The cations and radicals in CTQ #1 and #2 are called vinyl cations and vinyl radicals , respectively. Based on the regioselectivities reported in Model 1, what effect do alkyl substituents at the charged/radical carbon have on the energies of these species? Is this consistent with, or opposed to, their effect on R 3C+ and R 3C• species? 4. The geometry of a vinyl cation can be anticipated using VSEPR. Count electron domains in the cation below and predict all of the HCC bond angles. -

Nucleophilic Aromatic Substitution
NUCLEOPHILIC AROMATIC SUBSTITUTION Ms. Prerana Sanas M.Pharm (Pharmceutical Chemistry) Asst.Professor, NCRD’s Sterling Institute of Pharmacy Nerul Navi Mumbai Nucleophilic aromatic substitution results in the substitution of a halogen X on a benzene ring by a nucleophile (:Nu– ). Aryl halides undergo a limited number of substitution reactions with strong nucleophiles. NAS occurs by two mechanisms i) Bimoleccular displacement (Addition –Elimination) ii) Benzyne Formation( Elimination –Addition) 7/5/2019 Ms.Prerana Sanas 2 Bimolecular displacement (Addition – Elimination) Aryl halides with strong electron-withdrawing groups (such as NO2) on the ortho or para positions react with nucleophiles to afford substitution products. For example, treatment of p-chloronitrobenzene with hydroxide (– OH) affords p-nitrophenol by replacement of Cl by OH. Nucleophilic aromatic substitution occurs with a variety of strong nucleophiles, including – OH, – OR, – NH2, – SR, and in some cases, neutral nucleophiles such as NH3 and RNH2 . 7/5/2019 Ms.Prerana Sanas 3 Mechanism…… The mechanism of these reactions has two steps: Step i) Addition of the nucleophile (:Nu– ) forms a resonance-stabilized carbanion with a new C – Nu bond—three resonance structures can be drawn. • Step [1] is rate-determining since the aromaticity of the benzene ring is lost. In Step ii) loss of the leaving group re-forms the aromatic ring. This step is fast because the aromaticity of the benzene ring is restored. 7/5/2019 Ms.Prerana Sanas 4 Factors affecting Bimolecular displacement Increasing the number of electron-withdrawing groups increases the reactivity of the aryl halide. Electron-withdrawing groups stabilize the intermediate carbanion, and by the Hammond postulate, lower the energy of the transition state that forms it. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Lecture 5 Diastereoselective Addition Into Carbonyl Compounds
Lecture 5 Diastereoselective Addition into Carbonyl Compounds Containing α-Stereogenic Centres Learning outcomes: by the end of this lecture, and after answering the associated problems, you will be able to: 1. use the Felkin-Anh T.S. to predict the stereochemical outcome of reactions carried out on carbonyl compounds that possess an α-stereogenic centre; 2. rationalise the preferential adoption of a Felkin-Anh T.S. in nucleophilic addition reactions on steric and stereoelectronic grounds; 3. understand how the presence of an α-electron-withdrawing substituent affects the Felkin-Anh T.S.; 4. use the Felkin-Anh T.S. to prepare 1,2-syn diols from α-alkoxy ketones; 5. use the Cram chelation model to prepare 1,2-anti diols from α-alkoxy ketones. Stereoselective Addition of Nucleophiles into Ketones and Aldehydes containing α- Stereogenic Centres The addition of a nucleophile into a chiral ketone or aldehyde provides diastereoisomers. When the stereogenic centres in the substrate are close to the reacting carbonyl group (e.g. 1,2-disposed), then it is often possible to exploit this stereochemical information to control the stereoselectivity of the addition reaction. This method for controlling the stereochemical outcome of a reaction is known as substrate control. A number of models have been developed for predicting the stereochemical outcome of this type of reaction. Felkin-Anh Model Consider a carbonyl compound containing an α-stereogenic centre in which the three substituents at the α-site are well differentiated in size: O HO Nu Nu OH RL Nu RL RL R R R S M S M S RM R R R R R RS = small substituent RM = medium-sized substituent RL = large substituent Of the two diastereoisomeric alcohol addition products, one will be formed to a greater extent than the other. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC AROMATIC SUBSTITUTION by JAMES ERVIN SCHAMMERHORN III Associates of Science Murray
NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC AROMATIC SUBSTITUTION By JAMES ERVIN SCHAMMERHORN III Associates of Science Murray State College Tishomingo, Oklahoma 2003 Bachelor of Science in Chemistry Oklahoma State University Stillwater, Oklahoma 2006 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY May, 2011 1 NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC ARMOMATIC SUBSTITUTION Thesis Approved: Dr. Richard Bunce Thesis Adviser Dr. K. Darrell Berlin Dr. Ziad El-Rassi Dr. Nicholas Materer Dr. Andrew Mort Dr. Mark E. Payton Dean of the Graduate College ii ACKNOWLEDGMENTS I would like to express my sincere gratitude to Dr. R. A. Bunce, my research advisor, my graduate committee chairman and my friend. I have learned many valuable lessons working alongside him in his research laboratory. His ability to teach both organic theory and laboratory techniques have been instrumental in my research at Oklahoma State University. I am immensely thankful for his support, guidance and patience to me during the course of this research. His unique sense of humor always makes me laugh and makes it a joy to come to lab. I would also like to thank my committee members, Drs. K. D. Berlin, Z. El Rassi, N. F. Materer, and A. J. Mort their acceptance to be on my committee. Their insights into research and graduate life have been invaluable during my graduate career. I am especially grateful to Dr. Berlin for sharing his wealth of organic chemistry knowledge with me during the course of my research. Also, to Dr. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Ketenes 25/01/2014 Part 1
Baran Group Meeting Hai Dao Ketenes 25/01/2014 Part 1. Introduction Ph Ph n H Pr3N C A brief history Cl C Ph + nPr NHCl Ph O 3 1828: Synthesis of urea = the starting point of modern organic chemistry. O 1901: Wedekind's proposal for the formation of ketene equivalent (confirmed by Staudinger 1911) Wedekind's proposal (1901) 1902: Wolff rearrangement, Wolff, L. Liebigs Ann. Chem. 1902, 325, 129. 2 Wolff adopt a ketene structure in 1912. R 2 hν R R2 1905: First synthesis and characterization of a ketene: in an efford to synthesize radical 2, 1 ROH R C Staudinger has synthesized diphenylketene 3, Staudinger, H. et al., Chem. Ber. 1905, 1735. N2 1 RO CH or Δ C R C R1 1907-8: synthesis and dicussion about structure of the parent ketene, Wilsmore, O O J. Am. Chem. Soc. 1907, 1938; Wilsmore and Stewart Chem. Ber. 1908, 1025; Staudinger and Wolff rearrangement (1902) O Klever Chem. Ber. 1908, 1516. Ph Ph Cl Zn Ph O hot Pt wire Zn Br Cl Cl CH CH2 Ph C C vs. C Br C Ph Ph HO O O O O O O O 1 3 (isolated) 2 Wilsmore's synthesis and proposal (1907-8) Staudinger's synthesis and proposal (1908) wanted to make Staudinger's discovery (1905) Latest books: ketene (Tidwell, 1995), ketene II (Tidwell, 2006), Science of Synthesis, Vol. 23 (2006); Latest review: new direactions in ketene chemistry: the land of opportunity (Tidwell et al., Eur. J. Org. Chem. 2012, 1081). Search for ketenes, Google gave 406,000 (vs. -

Reaction Mechanism Lecture 3 : Nucleophilic And
Module 10 : Reaction mechanism Lecture 3 : Nucleophilic and Electrophilic Addition and Abstraction Objectives In this lecture you will learn the following Ligand activation by metal that leads to a direct external attack at the ligand. Nucleophilic addition and nucleophilic abstraction reactions. Electrophilic addition and electrophilic abstraction reactions. The nucleophilic and electrophilic substitution and abstraction reactions can be viewed as ways of activation of substrates to allow an external reagent to directly attack the metal activated ligand without requiring prior binding of the external reagent to the metal. The attacking reagent may be a nucleophile or an electrophile. The nucleophilic attack of the external reagent is favored if the LnM fragment is a poor π−base and a good σ−acid i.e., when the complex is cationic and/or when the other metal bound ligands are electron withdrawing such that the ligand getting activated gets depleted of electron density and can undergo an external attack by a nucleophile Nu−, like LiMe or OH−. The attack of the nucleophiles may result in the formation of a bond between the nucleophiles and the activated unsaturated substrate, in which case it is called nucleophilic addition, or may result in an abstraction of a part or the whole of the activated ligand, in which case it is called the nucleophilic abstraction. The nucleophilic addition and the abstraction reactions are discussed below. Nucleophilic addition An example of a nucleophilic addition reaction is shown below. Carbon monoxide (CO) as a ligand can undergo nucleophilic attack when bound to a metal center of poor π−basicity, as the carbon center of the CO ligand is electron deficient owing to the ligand to metal σ−donation not being fully compensated by the metal to ligand π−back donation. -
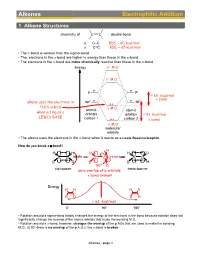
Alkenes Electrophilic Addition
Alkenes Electrophilic Addition 1 Alkene Structures chemistry of C C double bond σ C–C BDE ~ 80 kcal/mol π C=C BDE ~ 65 kcal/mol • The p-bond is weaker than the sigma-bond • The, electrons in the p-bond are higher in energy than those in the s-bond • The electrons in the p-bond are more chemically reactive than those in the s-bond Energy σ∗ M.O. π∗ M.O. p p ~ 65 kcal/mol 2 π bond alkene uses the electrons in sp2 sp THIS orbital π M.O. atomic atomic when acting as a orbitals orbitals ~ 81 kcal/mol LEWIS BASE carbon 1 carbon 2 σ bond σ M.O. molecular orbitals • The alkene uses the electrons in the p-bond when it reacts as a Lewis Base/nucleophile How do you break a p-bond? H H H H H D C C rotate C C rotate C C D D D D H 90° D cis-isomer trans-isomer zero overlap of p orbitals: π bond broken! Energy H H H D D D D H ~ 63 kcal/mol 0° 90° 180° • Rotation around a sigma-bond hardly changes the energy of the electrons in the bond because rotation does not significantly change the overlap of the atomic orbitals that make the bonding M.O. • Rotation around a p-bond, however, changes the overlap of the p AOs that are used to make the bonding M.O., at 90° there is no overlap of the p A.O.s, the p-bond is broken Alkenes : page 1 Distinguishing isomers trans- cis- • By now we are very familiar with cis- and trans-stereoisomers (diastereomers) • But what about, the following two structures, they can NOT be assigned as cis- or trans-, yet they are definitely stereoisomers (diastereomers), the directions in which their atoms point in space are different cis- / trans- Br Br We Need a different system to distinguish stereoisomers for C=C double bonds: Use Z/E notation.