Subject Index
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

A Macroscopic Demonstration of the Malaprade Reaction † † † † E
Demonstration Cite This: J. Chem. Educ. 2019, 96, 1199−1204 pubs.acs.org/jchemeduc How To Shrink Paper Money: A Macroscopic Demonstration of the Malaprade Reaction † † † † E. Brandon Strong, Brittany A. Lore, Emily R. Christensen, Nathaniel W. Martinez, ‡ and Andres W. Martinez*, † ‡ Department of Biological Sciences and Department of Chemistry & Biochemistry, California Polytechnic State University, San Luis Obispo, California 93401, United States *S Supporting Information ABSTRACT: Shrinking paper money is a captivating and memorable demonstration of chemistry in action that can be carried out using minimal equipment and reagents. Paper bills can be shrunk down to ∼25% of their initial surface area by treating them with an aqueous solution of sodium periodate. The cellulose in the paper bill is oxidized to dialdehyde cellulose via the Malaprade reaction, and the resulting change in size of the paper money provides macroscopic evidence of the reaction. The chemical change that occurs in the cellulose can be confirmed by a Tollen’s test. KEYWORDS: General Public, Elementary/Middle-School Science, High School/Introductory Chemistry, First-Year Undergraduate/General, Second-Year Undergraduate, Demonstrations, Hands-On-Learning/Manipulatives, Alcohols, Aldehydes/Ketones, Oxidation/Reduction ■ INTRODUCTION In 1937, Jackson and Hudson reported that a piece of filter paper shrank to 25% of its original surface area upon reacting Paper money is arguably one of the most recognizable objects 14 in the world. Magicians, artists, and scientists have all with periodic acid. The shrinkage was later attributed to the capitalized on the popularity of banknotes by using them as reorganization of the dialdehyde cellulose chains into nonlinear props to capture the attention of the public. -

Open PS Thesis - Clara Capparelli
The Pennsylvania State University The Graduate School Department of Material Science and Engineering INFLUENCE OF CARBON SPACERS AND ALKYL PENDANT CHAINS ON THE STABILITY OF QUATERNARY AMMONIUM CATIONS FOR ANION EXCHANGE MEMBRANES A Thesis in Material Science and Engineering by Clara Capparelli 2015 Clara Capparelli Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science August 2015 The thesis of Clara Capparelli was reviewed and approved* by the following: Michael A. Hickner Associate Professor of Materials Science and Engineering Thesis Advisor James Runt Professor of Polymer Science T.C. Mike Chung Professor of Material Science and Engineering Suzanne Mohney Professor of Material Science and Engineering and Electrical Engineering Chair, Intercollege Graduate Degree Program in Material Science and Engineering *Signatures are on file in the Graduate School iii ABSTRACT Proton and anion exchange membranes are of great importance in the function of fuel cells, one of the most promising technologies for renewable energy conversion. Proton exchange membrane fuel cells (PEMFC) have been studied extensively in the past couple of decades, and there have been tremendous advances in the development of these systems, especially in industries such as automotive and portable power. Anion exchange membranes (AEM) have caught the attention of scientists because they would allow for the development of fuel cells without costly precious metal catalysts, among other advantages. Efforts are being made in developing long-lived and high performance AEMs for fuel cell applications. Primarily, the focus in AEM research has been membrane stability. It has been observed that AEMs are not as stable as the state-of-the-art NAFION® PEM and demonstrations of cell performance beyond 1000 hours is rare. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Alcohol Oxidation
Alcohol oxidation Alcohol oxidation is an important organic reaction. Primary alcohols (R-CH2-OH) can be oxidized either Mechanism of oxidation of primary alcohols to carboxylic acids via aldehydes and The indirect oxidation of aldehyde hydrates primary alcohols to carboxylic acids normally proceeds via the corresponding aldehyde, which is transformed via an aldehyde hydrate (R- CH(OH)2) by reaction with water. The oxidation of a primary alcohol at the aldehyde level is possible by performing the reaction in absence of water, so that no aldehyde hydrate can be formed. Contents Oxidation to aldehydes Oxidation to ketones Oxidation to carboxylic acids Diol oxidation References Oxidation to aldehydes Oxidation of alcohols to aldehydes is partial oxidation; aldehydes are further oxidized to carboxylic acids. Conditions required for making aldehydes are heat and distillation. In aldehyde formation, the temperature of the reaction should be kept above the boiling point of the aldehyde and below the boiling point of the alcohol. Reagents useful for the transformation of primary alcohols to aldehydes are normally also suitable for the oxidation of secondary alcohols to ketones. These include: Oxidation of alcohols to aldehydes and ketones Chromium-based reagents, such as Collins reagent (CrO3·Py2), PDC or PCC. Sulfonium species known as "activated DMSO" which can result from reaction of DMSO with electrophiles, such as oxalyl chloride (Swern oxidation), a carbodiimide (Pfitzner-Moffatt oxidation) or the complex SO3·Py (Parikh-Doering oxidation). Hypervalent iodine compounds, such as Dess-Martin periodinane or 2-Iodoxybenzoic acid. Catalytic TPAP in presence of excess of NMO (Ley oxidation). Catalytic TEMPO in presence of excess bleach (NaOCl) (Oxoammonium-catalyzed oxidation). -

Reaction Mechanism Lecture 3 : Nucleophilic And
Module 10 : Reaction mechanism Lecture 3 : Nucleophilic and Electrophilic Addition and Abstraction Objectives In this lecture you will learn the following Ligand activation by metal that leads to a direct external attack at the ligand. Nucleophilic addition and nucleophilic abstraction reactions. Electrophilic addition and electrophilic abstraction reactions. The nucleophilic and electrophilic substitution and abstraction reactions can be viewed as ways of activation of substrates to allow an external reagent to directly attack the metal activated ligand without requiring prior binding of the external reagent to the metal. The attacking reagent may be a nucleophile or an electrophile. The nucleophilic attack of the external reagent is favored if the LnM fragment is a poor π−base and a good σ−acid i.e., when the complex is cationic and/or when the other metal bound ligands are electron withdrawing such that the ligand getting activated gets depleted of electron density and can undergo an external attack by a nucleophile Nu−, like LiMe or OH−. The attack of the nucleophiles may result in the formation of a bond between the nucleophiles and the activated unsaturated substrate, in which case it is called nucleophilic addition, or may result in an abstraction of a part or the whole of the activated ligand, in which case it is called the nucleophilic abstraction. The nucleophilic addition and the abstraction reactions are discussed below. Nucleophilic addition An example of a nucleophilic addition reaction is shown below. Carbon monoxide (CO) as a ligand can undergo nucleophilic attack when bound to a metal center of poor π−basicity, as the carbon center of the CO ligand is electron deficient owing to the ligand to metal σ−donation not being fully compensated by the metal to ligand π−back donation. -
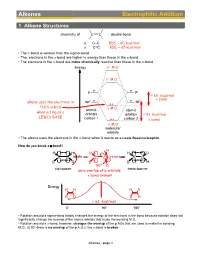
Alkenes Electrophilic Addition
Alkenes Electrophilic Addition 1 Alkene Structures chemistry of C C double bond σ C–C BDE ~ 80 kcal/mol π C=C BDE ~ 65 kcal/mol • The p-bond is weaker than the sigma-bond • The, electrons in the p-bond are higher in energy than those in the s-bond • The electrons in the p-bond are more chemically reactive than those in the s-bond Energy σ∗ M.O. π∗ M.O. p p ~ 65 kcal/mol 2 π bond alkene uses the electrons in sp2 sp THIS orbital π M.O. atomic atomic when acting as a orbitals orbitals ~ 81 kcal/mol LEWIS BASE carbon 1 carbon 2 σ bond σ M.O. molecular orbitals • The alkene uses the electrons in the p-bond when it reacts as a Lewis Base/nucleophile How do you break a p-bond? H H H H H D C C rotate C C rotate C C D D D D H 90° D cis-isomer trans-isomer zero overlap of p orbitals: π bond broken! Energy H H H D D D D H ~ 63 kcal/mol 0° 90° 180° • Rotation around a sigma-bond hardly changes the energy of the electrons in the bond because rotation does not significantly change the overlap of the atomic orbitals that make the bonding M.O. • Rotation around a p-bond, however, changes the overlap of the p AOs that are used to make the bonding M.O., at 90° there is no overlap of the p A.O.s, the p-bond is broken Alkenes : page 1 Distinguishing isomers trans- cis- • By now we are very familiar with cis- and trans-stereoisomers (diastereomers) • But what about, the following two structures, they can NOT be assigned as cis- or trans-, yet they are definitely stereoisomers (diastereomers), the directions in which their atoms point in space are different cis- / trans- Br Br We Need a different system to distinguish stereoisomers for C=C double bonds: Use Z/E notation. -

Solvating Alkylamine Hofmann Elimination in Zeolites Through Cooperative Adsorption Han Chen† and Omar A
Solvating Alkylamine Hofmann Elimination in Zeolites Through Cooperative Adsorption Han Chen† and Omar A. Abdelrahman †,‡* † Department of Chemical Engineering, University of Massachusetts Amherst, 686 N. Pleasant Street, Amherst, MA 01003, USA ‡ Catalysis Center for Energy Innovation, University of Delaware, 150 Academy Street, Newark, DE 19716, USA *Corresponding Author: [email protected] Abstract. A kinetic investigation of the vapor phase Hofmann elimination of tert-butylamine over H- ZSM-5 reveals a carbocation mediated E1-like mechanism, where isobutene and ammonia are exclusively produced over Brønsted acid sites. Hofmann elimination kinetics are found to be insensitive to Al content or siting, varying only with alkylamine carbocation stability (rtertiary > rsecondary > rprimary). Under conditions of complete tert-butylamine surface coverage, experimentally measurable apparent kinetics are directly equivalent to the intrinsic kinetics of the rate determining unimolecular surface elimination. The direct measurement of elementary step kinetics served as a water-free reactive probe, providing a direct measurement of the impact of water on solid Brønsted acid catalyzed chemistries at a microscopic level. Over a range of temperatures (453‒513 K) and tert-butylamine partial pressures (6.8×10-2‒6.8 kPa), water reversibly inhibits the rate of Hofmann elimination. Despite expected changes in aluminosilicate hydrophobicity, the water-induced inhibition is found to be insensitive to Al content, demonstrated to be due to one water molecule per Brønsted acid site. Regardless of the significant reduction in the rate of Hofmann elimination, kinetic interrogations and operando spectroscopic measurements reveal that the coverage of TBA adsorbed on H-ZSM-5 is unaltered in the presence of water. -

Heterocyclic Chemistrychemistry
HeterocyclicHeterocyclic ChemistryChemistry Professor J. Stephen Clark Room C4-04 Email: [email protected] 2011 –2012 1 http://www.chem.gla.ac.uk/staff/stephenc/UndergraduateTeaching.html Recommended Reading • Heterocyclic Chemistry – J. A. Joule, K. Mills and G. F. Smith • Heterocyclic Chemistry (Oxford Primer Series) – T. Gilchrist • Aromatic Heterocyclic Chemistry – D. T. Davies 2 Course Summary Introduction • Definition of terms and classification of heterocycles • Functional group chemistry: imines, enamines, acetals, enols, and sulfur-containing groups Intermediates used for the construction of aromatic heterocycles • Synthesis of aromatic heterocycles • Carbon–heteroatom bond formation and choice of oxidation state • Examples of commonly used strategies for heterocycle synthesis Pyridines • General properties, electronic structure • Synthesis of pyridines • Electrophilic substitution of pyridines • Nucleophilic substitution of pyridines • Metallation of pyridines Pyridine derivatives • Structure and reactivity of oxy-pyridines, alkyl pyridines, pyridinium salts, and pyridine N-oxides Quinolines and isoquinolines • General properties and reactivity compared to pyridine • Electrophilic and nucleophilic substitution quinolines and isoquinolines 3 • General methods used for the synthesis of quinolines and isoquinolines Course Summary (cont) Five-membered aromatic heterocycles • General properties, structure and reactivity of pyrroles, furans and thiophenes • Methods and strategies for the synthesis of five-membered heteroaromatics -

Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-Species
Molecules 2014, 19, 6534-6582; doi:10.3390/molecules19056534 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-species Vincenzo Piccialli Dipartimento di Scienze Chimiche, Università degli Studi di Napoli ―Federico II‖, Via Cintia 4, 80126, Napoli, Italy; E-Mail: [email protected]; Tel.: +39-081-674111; Fax: +39-081-674393 Received: 24 February 2014; in revised form: 14 May 2014 / Accepted: 16 May 2014 / Published: 21 May 2014 Abstract: In the last years ruthenium tetroxide is increasingly being used in organic synthesis. Thanks to the fine tuning of the reaction conditions, including pH control of the medium and the use of a wider range of co-oxidants, this species has proven to be a reagent able to catalyse useful synthetic transformations which are either a valuable alternative to established methods or even, in some cases, the method of choice. Protocols for oxidation of hydrocarbons, oxidative cleavage of C–C double bonds, even stopping the process at the aldehyde stage, oxidative cleavage of terminal and internal alkynes, oxidation of alcohols to carboxylic acids, dihydroxylation of alkenes, oxidative degradation of phenyl and other heteroaromatic nuclei, oxidative cyclization of dienes, have now reached a good level of improvement and are more and more included into complex synthetic sequences. The perruthenate ion is a ruthenium (VII) oxo-species. Since its introduction in the mid-eighties, tetrapropylammonium perruthenate (TPAP) has reached a great popularity among organic chemists and it is mostly employed in catalytic amounts in conjunction with N-methylmorpholine N-oxide (NMO) for the mild oxidation of primary and secondary alcohols to carbonyl compounds. -

Organic Seminar Abstracts
L I B RA R.Y OF THE UN IVE.R.SITY Of ILLI NOIS 547 l£6s \ 954/55 PV Return this book on or before the Latest Date stamped below. University of Illinois Library Llf.1—H41 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/organicsemi195455univ \7^ — SEMINAR TOPICS CHEMISTRY 435 I Semester 1954-55 The Quinolizinium Ion and Some of its Derivatives Harvey M. Loux, September 24 1 The Stereochemistry of Atropine and Cocaine Ho E . Knipmeyer, September 24 4 Recent Developments in the Chemistry of Cinnoline Derivatives Roger H. Kottke , October 1 8 Interpretation of Electrophiiic Aromatic Substitution and Solvolysis of Allylic and Benzhydryl Chlorides in Terms of Hyperconjugation Robert D. Stolow, October 1 11 The Decahydronaphtholic Acids and Their Relationship to the Decalols and the Decalylamines Robert J. Harder, October 8 14 Bis-Cyclopentadienyl Metal Compounds Edwin L. DeYoung, October 8 17 Ion-Pairs as Intermediates in Solvolysis Reactions Arthur H. Goldkamp, October 15 21 Synthesis of Morphine Mohan D. Nair, October 15 24 Structural and Geometrical Isomerism in the Oxidation of Azo Compounds D. F. Morrow, October 22 27 Stereochemical Aspects of Thermochromism John W. Johnson, Jr., October 22 30 A New Method for the Preparation of Olefins --The Pyrolysis of Sulfites F. M. Scheidt, October 29 33 Recent Studies of Macrocyclic Ring Systems: Cyclophanes Fred P. Hauck, Jr., October 29 36 Mechanism of the Para-Claisen Rearrangement Hugh H . Gibbs , November 5 40 A Proposed Mechanism for Basic c is -Dehydrohalogenat ion Robert M.