Nucleophilic Aromatic Substitution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development and Application of Metal-Catalyzed Processes for Organic Synthesis
The Development and Application of Metal-Catalyzed Processes for Organic Synthesis by Edward J. Hennessy B.A. Chemistry Northwestern University, 2000 Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN ORGANIC CHEMISTRY at the Massachusetts Institute of Technology June 2005 © 2005 Massachusetts Institute of Technology All Rights Reserved Signature of Author: " - - 1_"' .Da-uirnt of Chemistry May 19, 2005 Certified by: Stephen L. Buchwald Camille Dreyfus Professor of Chemistry Thesis Supervisor Accepted by: Robert W. Field Chairman, Departmental Committee on Graduate Students ARCHIVES: This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: -/ Professor Gregory C. Fu: " Ch Chair Professor Stephen L. Buchwald Thesis Supervisor Professor Timothy M. Swager: , -L i \ O 2 The Development and Application of Metal-Catalyzed Processes for Organic Synthesis by Edward J. Hennessy Submitted to the Department of Chemistry on May 19, 2005 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the Massachusetts Institute of Technology ABSTRACT Chapter 1. Copper-Catalyzed Arylation of Stabilized Carbanions A mild, general catalytic system for the synthesis of a-aryl malonates has been developed. Aryl iodides bearing a variety of functional groups can be effectively coupled to diethyl malonate in high yields using inexpensive and widely available reagents, making this a superior method to those previously described that employ copper reagents or catalysts. The functional group tolerance of the process developed makes it complementary to analogous palladium-catalyzed couplings. Importantly, a set of mild reaction conditions has been developed that minimize product decomposition, a problem that had not been addressed previously in the literature. -

The Total Synthesis of Securinine and Other Methodology Studies
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2010 The total synthesis of securinine and other methodology studies Bhartesh Dhudshia University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Dhudshia, Bhartesh, "The total synthesis of securinine and other methodology studies" (2010). Electronic Theses and Dissertations. 8275. https://scholar.uwindsor.ca/etd/8275 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. The Total Synthesis of Securinine and Other Methodology Studies by Bhartesh Dhudshia A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements -

Organic Synthesis: New Vistas in the Brazilian Landscape
Anais da Academia Brasileira de Ciências (2018) 90(1 Suppl. 1): 895-941 (Annals of the Brazilian Academy of Sciences) Printed version ISSN 0001-3765 / Online version ISSN 1678-2690 http://dx.doi.org/10.1590/0001-3765201820170564 www.scielo.br/aabc | www.fb.com/aabcjournal Organic Synthesis: New Vistas in the Brazilian Landscape RONALDO A. PILLI and FRANCISCO F. DE ASSIS Instituto de Química, UNICAMP, Rua José de Castro, s/n, 13083-970 Campinas, SP, Brazil Manuscript received on September 11, 2017; accepted for publication on December 29, 2017 ABSTRACT In this overview, we present our analysis of the future of organic synthesis in Brazil, a highly innovative and strategic area of research which underpins our social and economical progress. Several different topics (automation, catalysis, green chemistry, scalability, methodological studies and total syntheses) were considered to hold promise for the future advance of chemical sciences in Brazil. In order to put it in perspective, contributions from Brazilian laboratories were selected by the citations received and importance for the field and were benchmarked against some of the most important results disclosed by authors worldwide. The picture that emerged reveals a thriving area of research, with new generations of well-trained and productive chemists engaged particularly in the areas of green chemistry and catalysis. In order to fulfill the promise of delivering more efficient and sustainable processes, an integration of the academic and industrial research agendas is to be expected. On the other hand, academic research in automation of chemical processes, a well established topic of investigation in industrial settings, has just recently began in Brazil and more academic laboratories are lining up to contribute. -

Lecture 5 Diastereoselective Addition Into Carbonyl Compounds
Lecture 5 Diastereoselective Addition into Carbonyl Compounds Containing α-Stereogenic Centres Learning outcomes: by the end of this lecture, and after answering the associated problems, you will be able to: 1. use the Felkin-Anh T.S. to predict the stereochemical outcome of reactions carried out on carbonyl compounds that possess an α-stereogenic centre; 2. rationalise the preferential adoption of a Felkin-Anh T.S. in nucleophilic addition reactions on steric and stereoelectronic grounds; 3. understand how the presence of an α-electron-withdrawing substituent affects the Felkin-Anh T.S.; 4. use the Felkin-Anh T.S. to prepare 1,2-syn diols from α-alkoxy ketones; 5. use the Cram chelation model to prepare 1,2-anti diols from α-alkoxy ketones. Stereoselective Addition of Nucleophiles into Ketones and Aldehydes containing α- Stereogenic Centres The addition of a nucleophile into a chiral ketone or aldehyde provides diastereoisomers. When the stereogenic centres in the substrate are close to the reacting carbonyl group (e.g. 1,2-disposed), then it is often possible to exploit this stereochemical information to control the stereoselectivity of the addition reaction. This method for controlling the stereochemical outcome of a reaction is known as substrate control. A number of models have been developed for predicting the stereochemical outcome of this type of reaction. Felkin-Anh Model Consider a carbonyl compound containing an α-stereogenic centre in which the three substituents at the α-site are well differentiated in size: O HO Nu Nu OH RL Nu RL RL R R R S M S M S RM R R R R R RS = small substituent RM = medium-sized substituent RL = large substituent Of the two diastereoisomeric alcohol addition products, one will be formed to a greater extent than the other. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

Ketenes 25/01/2014 Part 1
Baran Group Meeting Hai Dao Ketenes 25/01/2014 Part 1. Introduction Ph Ph n H Pr3N C A brief history Cl C Ph + nPr NHCl Ph O 3 1828: Synthesis of urea = the starting point of modern organic chemistry. O 1901: Wedekind's proposal for the formation of ketene equivalent (confirmed by Staudinger 1911) Wedekind's proposal (1901) 1902: Wolff rearrangement, Wolff, L. Liebigs Ann. Chem. 1902, 325, 129. 2 Wolff adopt a ketene structure in 1912. R 2 hν R R2 1905: First synthesis and characterization of a ketene: in an efford to synthesize radical 2, 1 ROH R C Staudinger has synthesized diphenylketene 3, Staudinger, H. et al., Chem. Ber. 1905, 1735. N2 1 RO CH or Δ C R C R1 1907-8: synthesis and dicussion about structure of the parent ketene, Wilsmore, O O J. Am. Chem. Soc. 1907, 1938; Wilsmore and Stewart Chem. Ber. 1908, 1025; Staudinger and Wolff rearrangement (1902) O Klever Chem. Ber. 1908, 1516. Ph Ph Cl Zn Ph O hot Pt wire Zn Br Cl Cl CH CH2 Ph C C vs. C Br C Ph Ph HO O O O O O O O 1 3 (isolated) 2 Wilsmore's synthesis and proposal (1907-8) Staudinger's synthesis and proposal (1908) wanted to make Staudinger's discovery (1905) Latest books: ketene (Tidwell, 1995), ketene II (Tidwell, 2006), Science of Synthesis, Vol. 23 (2006); Latest review: new direactions in ketene chemistry: the land of opportunity (Tidwell et al., Eur. J. Org. Chem. 2012, 1081). Search for ketenes, Google gave 406,000 (vs. -

Aryl Halides
Block 3 Aromatic Hydrocarbons and Halogen Derivatives UNIT14 ARYL HALIDES Structure 14.1 Introduction Electophilic Substitution Reactions Expected Learning Outcomes 14.2 Structure and Reactivity Reactions due to C−X bond 14.5 Reactivity and Relative 14.3 Preparation of Aryl Halides Strength of C−X Bonds in 14.4 Reaction of Aryl Halides Halogen Derivatives Nucleophilic Substitution by 14.6 Summary Addition-Elimination 14.7 Terminal Questions Nucleophilic Substitution via 14.8 Answers Benzene Intermediate 14.1 INTRODUCTION In Unit 13, we have pointed out that there is a difference in the nature of C−X bond of aryl halides and aryl halides. Because of this aryl halides differ from the alkyl halides in their preparation and properties. In this Unit, we will study the unique chemistry of aryl halides. First, we shall take up the structure and reactivity of aryl halides, which is followed by their preparations and properties. At the end of the unit we shall compare the reactivity and relative strength of C−X bond in different type of halogen derivatives. Expected Learning Outcomes After studying this unit, you should be able to: explain why aryl halides are less reactive than alkyl halides, outline the methods of preparation of aryl halides, describe the reactions of aryl halides, and explain the difference in structure and reactivity of alkyl, alkenyl and aryl halides towards nucleophilic substitution reactions. 94 Unit 14 Aryl Halides 14.2 STRUCTURE AND REACTIVITY Before going into the details of the preparations and properties of aryl halides let us take a look at the structure and reactivity of these compounds so that we can understand why their reactions are different from alkyl halides. -

Aromatic Substitution Reactions: an Overview
Review Article Published: 03 Feb, 2020 SF Journal of Pharmaceutical and Analytical Chemistry Aromatic Substitution Reactions: An Overview Kapoor Y1 and Kumar K1,2* 1School of Pharmaceutical Sciences, Apeejay Stya University, Sohna-Palwal Road, Sohna, Gurgaon, Haryana, India 2School of Pharmacy and Technology Management, SVKM’s NMIMS University, Hyderabad, Telangana, India Abstract The introduction or replacement of substituent’s on aromatic rings by substitution reactions is one of the most fundamental transformations in organic chemistry. On the basis of the reaction mechanism, these substitution reactions can be divided into electrophilic, nucleophilic, radical, and transition metal catalyzed. This article also focuses on electrophilic and nucleophilic substitution mechanisms. Introduction The replacement of an atom, generally hydrogen, or a group attached to the carbon from the benzene ring by another group is known as aromatic substitution. The regioselectivity of these reactions depends upon the nature of the existing substituent and can be ortho, Meta or Para selective. Electrophilic Aromatic Substitution (EAS) reactions are important for synthetic purposes and are also among the most thoroughly studied classes of organic reactions from a mechanistic point of view. A wide variety of electrophiles can effect aromatic substitution. Usually, it is a substitution of some other group for hydrogen that is of interest, but this is not always the case. For example, both silicon and mercury substituent’s can be replaced by electrophiles. The reactivity of a particular electrophile determines which aromatic compounds can be successfully substituted. Despite the wide range of electrophilic species and aromatic ring systems that can undergo substitution, a single broad mechanistic picture encompasses most EAS reactions. -

Aromatic Nucleophilic Substitution Reaction
Aromatic Nucleophilic Substitution Reaction DR. RAJENDRA R TAYADE ASSISTANT PROFESSOR DEPARTMENT OF CHEMISTRY INSTITUTE OF SCIENCE, NAGPUR Principles There are four principal mechanisms for aromatic nucleophilic substitution which are similar to that of aliphatic nucleophilic substitution. (SN1, SN2, SNi, SET Mechanism) 1. SNAr Mechanism- addition / elimination CF3, CN, CHO, COR, COOH, Br, Cl, I Common Activating Groups for NAS Step [1] Addition of the nucleophile (:Nu–) to form a carbanion Addition of the nucleophile (:Nu–) forms a resonance-stabilized carbanion with a new C – Nu bond— three resonance structures can be drawn. • Step is rate-determining • Aromaticity of the benzene ring is lost Step [2] loss of the leaving group re-forms the aromatic ring. • This step is fast because the aromaticity of the benzene ring is restored. ? Explain why a methoxy group (CH3O) increases the rate of electrophilic aromatic substitution, but decreases the rate of nucleophilic aromatic substitution. 2.ArSN1 Mechanism- elimination /addition • This mechanism operates in the reaction of diazonium salts with nucleophiles. •The driving force resides in the strength of the bonding in the nitrogen molecule that makes it a particularly good leaving group. 3.Benzyne Mechanism- elimination /addition Step [1] Elimination of HX to form benzyne Elimination of H and X from two adjacent carbons forms a reactive benzyne intermediate Step [2] Nucleophilic addition to form the substitution product Addition of the nucleophile (–OH in this case) and protonation form the substitution product Evidence for the Benzyne Mechanism Trapping in Diels/Alder Reaction O O B E N Z Y N E C C O O O D i e l s / A l d e r O N H 3 N N Dienophile Diene A d d u c t Substrate Modification – absence of a hydrogens LG Substituent Substituent No Reaction Base Isotopic Labeling LG Nu H Nu Structure of Benzyne • The σ bond is formed by overlap of two sp2 hybrid orbitals. -

Simple, Efficient Catalyst System for the Palladium-Catalyzed Amination of Aryl Chlorides, Bromides, and Triflates
1158 J. Org. Chem. 2000, 65, 1158-1174 Simple, Efficient Catalyst System for the Palladium-Catalyzed Amination of Aryl Chlorides, Bromides, and Triflates John P. Wolfe,† Hiroshi Tomori, Joseph P. Sadighi,‡ Jingjun Yin, and Stephen L. Buchwald* Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 Received October 29, 1999 Palladium complexes supported by (o-biphenyl)P(t-Bu)2 (3)or(o-biphenyl)PCy2 (4) are efficient catalysts for the catalytic amination of a wide variety of aryl halides and triflates. Use of ligand 3 allows for the room-temperature catalytic amination of many aryl chloride, bromide, and triflate substrates, while ligand 4 is effective for the amination of functionalized substrates or reactions of acyclic secondary amines. The catalysts perform well for a large number of different substrate combinations at 80-110 °C, including chloropyridines and functionalized aryl halides and triflates using 0.5-1.0 mol % Pd; some reactions proceed efficiently at low catalyst levels (0.05 mol % Pd). These ligands are effective for almost all substrate combinations that have been previously reported with various other ligands, and they represent the most generally effective catalyst system reported to date. Ligands 3 and 4 are air-stable, crystalline solids that are commercially available. Their effectiveness is believed to be due to a combination of steric and electronic properties that promote oxidative addition, Pd-N bond formation, and reductive elimination. Owing to the many important applications of aniline derivatives, and the limitations of most methods for their synthesis, a considerable amount of effort has been recently devoted to the development of catalysts that are capable of effecting the cross-coupling of amines with aryl halides and sulfonates.1 However, the proper choice of catalyst (Pd source, ligand choice) is crucial for the success of these reactions. -
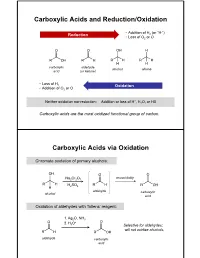
Carboxylic Acids and Reduction/Oxidation Carboxylic
Carboxylic Acids and Reduction/Oxidation • Addition of H (or “H-”) Reduction 2 • Loss of O2 or O carboxylic aldehyde alcohol alkane acid (or ketone) • Loss of H 2 Oxidation • Addition of O2 or O + Neither oxidation nor reduction: Addition or loss of H , H2O, or HX Carboxylic acids are the most oxidized functional group of carbon. Carboxylic Acids via Oxidation Chromate oxidation of primary alcohols: unavoidably Na2Cr2O7 H2SO4 aldehyde carboxylic alcohol acid Oxidation of aldehydes with Tollens’ reagent: 1. Ag2O, NH3 2. H O+ 3 Selective for aldehydes; will not oxidize alcohols. aldehyde carboxylic acid Carboxylic Acids via Oxidation Oxidative cleavage of alkynes: 1. O3 2. H2O internal alkyne Oxidation of benzylic carbons: KMnO4 Purifying Carboxylic Acids by Acid-Base Extraction Acidic and basic organic molecules can be separated from other substances by manipulating their protonation state and solubility. Let’s say we run a reaction. Na2Cr2O7 + unreacted H2SO4 + Na+ 3+ We want this… + Cr(H2O)6 + organic + HSO - …but not these. side products 4 How do we isolate our desired product from the other materials, without using distillation or chromatography? Acid-Base Extraction Step 1: Remove inorganics from organics via differential solubility. a separatory (sept) funnel - Na+ HSO4 Cr(H O) 3+ H2O 2 6 CHCl3 + organic side densities: products (H2O) = 1 g/mL (CHCl3) = 1.48 g/mL A large density difference ensures the two liquids separate completely and quickly. (Much faster than, say, oil and water.) Step 2: Add basic water to deprotonate carboxylic acid, and transfer it to the aqueous phase. - Na+ HSO4 3+ discard Cr(H2O)6 H2O layer + organic + organic side side products products add discard NaOH/ H2O CHCl3 layer + organic side products Step 3: Re-acidify carboxylate to return it to organic solution.