UNIT-III: Mechanistic and Sterochemical Aspects of Addition Reaction Involving Electrophiles & Nucleophiles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry Lesson 10 Reactions of Organic Molecules Combusition Reactions Substitution Reactions
Organic Chemistry Lesson 10 Reactions of Organic Molecules Combusition Reactions Substitution Reactions Physical Sciences Grade 12 1 Instructions Lesson 10 (16 April 2020) 1. Read ALL the slides in this document. 2. Homework: Just understand the theory for now. • If you have any questions, post them onto your WhatsApp group and your teacher will attend to them when (s)he has a chance. • Additional Resources: • Summary on pg.132 of your textbook. Physical Sciences Grade 12 2 Types of Reactions of Organic Compounds 1. Esterification: formation of an ester from an alcohol and a carboxylic acid. 2. Combustion Reactions (aka Oxidation): organic molecule + oxygen. 3. Substitution Reactions: one or more atoms are replaced by other atoms. (i) alkanes to alkyl halides; (ii) alcohols to alkyl halides; (iii) alkyl halides to alcohols 4. Addition Reactions: one or more atoms are added. (i) hydrogenation; (ii) halogenation; (iii) hydrohalogenation; (iv) hydration 5. Elimination Reactions: one or more atoms are removed. (i) dehydrohalogenation; (ii) dehydration; (iii) cracking of alkanes Physical Sciences Grade 12 3 1. Esterification • Already discussed (refer to earlier notes). Physical Sciences Grade 12 4 2. Combustion Reactions, i.e. Oxidation • A combustion reaction is the reaction of an organic molecule with oxygen. • Please note: “combustion” does NOT mean “explosion”. All combustion reactions release energy but most do so WITHOUT any explosion. • Alkanes – in the form of fossil fuels – are currently the main source of energy worldwide as their combustion is highly exothermic. • Examples: Burning wood for a braai. Propane combusting in a gas stove. Petrol combusting in a car engine. Physical Sciences Grade 12 5 2. -

Activity 33. Hydrohalogenation & Hydration of Alkynes
Chem 201, Fall 2010 Activity 33, M Nov 22 Activity 33. Hydrohalogenation & Hydration of Alkynes Alkynes undergo many of the same addition reactions that alkenes do, including additions that require carbocation intermediates. Different energy and geometry changes may be required for additions to an alkyne and an alkene so some surprising outcomes may occur with alkynes. Model 1. Addition of HX Hydrogen halides (HCl, HBr, HI) add to alkynes to make vinyl halides . A carbocation mechanism is followed when the reaction is performed in the dark in a peroxide-free solvent. This addition obeys Markovnikov’s rule. A free-radical-chain mechanism is followed when HBr addition is initiated by organic peroxides. This addition gives an anti-Markonikov product. Critical Thinking Questions 1. Draw the carbocations indicated by the two bouncy curved arrows (below). Based on the observation that the carbocation mechanism yields a Markovnikov product, circle the more stable carbocation. 2. (CI) Draw the free radicals indicated by the two competing pathways (below). Based on the observation that the free-radical mechanism yields an anti-Markovnikov product, circle the more stable free radical. 3. The cations and radicals in CTQ #1 and #2 are called vinyl cations and vinyl radicals , respectively. Based on the regioselectivities reported in Model 1, what effect do alkyl substituents at the charged/radical carbon have on the energies of these species? Is this consistent with, or opposed to, their effect on R 3C+ and R 3C• species? 4. The geometry of a vinyl cation can be anticipated using VSEPR. Count electron domains in the cation below and predict all of the HCC bond angles. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC AROMATIC SUBSTITUTION by JAMES ERVIN SCHAMMERHORN III Associates of Science Murray
NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC AROMATIC SUBSTITUTION By JAMES ERVIN SCHAMMERHORN III Associates of Science Murray State College Tishomingo, Oklahoma 2003 Bachelor of Science in Chemistry Oklahoma State University Stillwater, Oklahoma 2006 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY May, 2011 1 NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC ARMOMATIC SUBSTITUTION Thesis Approved: Dr. Richard Bunce Thesis Adviser Dr. K. Darrell Berlin Dr. Ziad El-Rassi Dr. Nicholas Materer Dr. Andrew Mort Dr. Mark E. Payton Dean of the Graduate College ii ACKNOWLEDGMENTS I would like to express my sincere gratitude to Dr. R. A. Bunce, my research advisor, my graduate committee chairman and my friend. I have learned many valuable lessons working alongside him in his research laboratory. His ability to teach both organic theory and laboratory techniques have been instrumental in my research at Oklahoma State University. I am immensely thankful for his support, guidance and patience to me during the course of this research. His unique sense of humor always makes me laugh and makes it a joy to come to lab. I would also like to thank my committee members, Drs. K. D. Berlin, Z. El Rassi, N. F. Materer, and A. J. Mort their acceptance to be on my committee. Their insights into research and graduate life have been invaluable during my graduate career. I am especially grateful to Dr. Berlin for sharing his wealth of organic chemistry knowledge with me during the course of my research. Also, to Dr. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

20 More About Oxidation–Reduction Reactions
More About 20 Oxidation–Reduction Reactions OOC n important group of organic reactions consists of those that O A involve the transfer of electrons C from one molecule to another. Organic chemists H OH use these reactions—called oxidation–reduction reactions or redox reactions—to synthesize a large O variety of compounds. Redox reactions are also important C in biological systems because many of these reactions produce HH energy. You have seen a number of oxidation and reduction reactions in other chapters, but discussing them as a group will give you the opportunity to CH3OH compare them. In an oxidation–reduction reaction, one compound loses electrons and one com- pound gains electrons. The compound that loses electrons is oxidized, and the one that gains electrons is reduced. One way to remember the difference between oxidation and reduction is with the phrase “LEO the lion says GER”: Loss of Electrons is Oxi- dation; Gain of Electrons is Reduction. The following is an example of an oxidation–reduction reaction involving inorganic reagents: Cu+ + Fe3+ ¡ Cu2+ + Fe2+ In this reaction,Cu+ loses an electron, so Cu+ is oxidized. Fe3+ gains an electron, so Fe3+ is reduced. The reaction demonstrates two important points about oxidation– reduction reactions. First, oxidation is always coupled with reduction. In other words, a compound cannot gain electrons (be reduced) unless another compound in the reaction simultaneously loses electrons (is oxidized). Second, the compound that is oxidized (Cu+) is called the reducing agent because it loses the electrons that are used to reduce the other compound (Fe3+). Similarly, the compound that is reduced (Fe3+) is called the oxidizing agent because it gains the electrons given up by the other compound (Cu+) when it is oxidized. -

Aromatic Substitution Reactions: an Overview
Review Article Published: 03 Feb, 2020 SF Journal of Pharmaceutical and Analytical Chemistry Aromatic Substitution Reactions: An Overview Kapoor Y1 and Kumar K1,2* 1School of Pharmaceutical Sciences, Apeejay Stya University, Sohna-Palwal Road, Sohna, Gurgaon, Haryana, India 2School of Pharmacy and Technology Management, SVKM’s NMIMS University, Hyderabad, Telangana, India Abstract The introduction or replacement of substituent’s on aromatic rings by substitution reactions is one of the most fundamental transformations in organic chemistry. On the basis of the reaction mechanism, these substitution reactions can be divided into electrophilic, nucleophilic, radical, and transition metal catalyzed. This article also focuses on electrophilic and nucleophilic substitution mechanisms. Introduction The replacement of an atom, generally hydrogen, or a group attached to the carbon from the benzene ring by another group is known as aromatic substitution. The regioselectivity of these reactions depends upon the nature of the existing substituent and can be ortho, Meta or Para selective. Electrophilic Aromatic Substitution (EAS) reactions are important for synthetic purposes and are also among the most thoroughly studied classes of organic reactions from a mechanistic point of view. A wide variety of electrophiles can effect aromatic substitution. Usually, it is a substitution of some other group for hydrogen that is of interest, but this is not always the case. For example, both silicon and mercury substituent’s can be replaced by electrophiles. The reactivity of a particular electrophile determines which aromatic compounds can be successfully substituted. Despite the wide range of electrophilic species and aromatic ring systems that can undergo substitution, a single broad mechanistic picture encompasses most EAS reactions. -
![Mechanism, Regioselectivity, and the Kinetics of Phosphine-Catalyzed [3+2] Cycloaddition Reactions of Allenoates and Electron-Deficient Alkenes](https://docslib.b-cdn.net/cover/6186/mechanism-regioselectivity-and-the-kinetics-of-phosphine-catalyzed-3-2-cycloaddition-reactions-of-allenoates-and-electron-deficient-alkenes-1186186.webp)
Mechanism, Regioselectivity, and the Kinetics of Phosphine-Catalyzed [3+2] Cycloaddition Reactions of Allenoates and Electron-Deficient Alkenes
FULL PAPER DOI: 10.1002/chem.200701725 Mechanism, Regioselectivity, and the Kinetics of Phosphine-Catalyzed [3+2] Cycloaddition Reactions of Allenoates and Electron-Deficient Alkenes Yong Liang,[a] Song Liu,[a] Yuanzhi Xia,[a, b] Yahong Li,[b, c] and Zhi-Xiang Yu*[a] Abstract: With the aid of computations alyzed [3+ 2] cycloaddition reaction of theories. Isotopic labeling experiments and experiments, the detailed mecha- allenoates and electron-deficient al- combined with DFT calculations nism of the phosphine-catalyzed [3+ 2] kenes. DFT calculations and FMO showed that the commonly accepted cycloaddition reactions of allenoates analysis revealed that an electron-with- intramolecular [1,2]-proton shift should and electron-deficient alkenes has been drawing group is required in the allene be corrected to a water-catalyzed [1,2]- investigated. It was found that this re- to ensure the generation of the 1,3- proton shift. Additional isotopic label- action includes four consecutive pro- dipole kinetically and thermodynami- ing experiments of the hetero-[3+2] cesses: 1) In situ generation of a 1,3- cally. Atoms-in-molecules (AIM) cycloaddition of allenoates and elec- dipole from allenoate and phosphine, theory was used to analyze the stability tron-deficient imines further support 2) stepwise [3+2] cycloaddition, 3) a of the 1,3-dipole. The regioselectivity this finding. This investigation has also water-catalyzed [1,2]-hydrogen shift, of the [3+2] cycloaddition can be ra- been extended to the study of the and 4) elimination of the phosphine tionalized very well by FMO and AIM phosphine-catalyzed [3+2] cycloaddi- catalyst. -
![Regioselectivity in the [2 + 2] Cyclo-Addition Reaction of Triplet Carbonyl Compounds to Substituted Alkenes](https://docslib.b-cdn.net/cover/2491/regioselectivity-in-the-2-2-cyclo-addition-reaction-of-triplet-carbonyl-compounds-to-substituted-alkenes-1292491.webp)
Regioselectivity in the [2 + 2] Cyclo-Addition Reaction of Triplet Carbonyl Compounds to Substituted Alkenes
J. Chem. Sci., Vol. 117, No. 5, September 2005, pp. 561–571. © Indian Academy of Sciences. Regioselectivity in the [2 + 2] cyclo-addition reaction of triplet carbonyl compounds to substituted alkenes (Paterno–Büchi reaction): A spin-polarized conceptual DFT approach B PINTÉR,1 F DE PROFT,2 T VESZPRÉMI1 and P GEERLINGS2,* 1Inorganic Chemistry Department, Budapest University of Technology and Economics (BUTE), Szent Gellért tér 4, H-1521, Budapest, Hungary 2Eenheid Algemene Chemie (ALGC), Vrije Universiteit Brussel (VUB), Faculteit, Wetenschappen, Pleinlaan 2, 1050 Brussels, Belgium e-mail: [email protected] Abstract. Regioselectivity of the photochemical [2 + 2] cyclo-addition of triplet carbonyl compounds with a series of ground state electron-rich and electron-poor alkenes, the Paterno–Büchi reaction, is studied. Activation barriers for the first step of the triplet reaction are computed in the case of the O-attack. Next, the observed regioselectivity is explained using a series of DFT-based reactivity indices. In the first step, we use the local softness and the local HSAB principle within a softness matching approach, and explain the relative activation barriers of the addition step. In the final step, the regioselectivity is assessed within the framework of spin-polarized conceptual density functional theory, considering response functions of the system’s external potential v, number of electrons N and spin number Ns, being the difference between the number of a and b electrons in the spin-polarized system. Although the concept of local spin philicity, in- troduced recently within this theory, appears less suited to predict the regioselectivity in this reaction, the correct regioselectivity emerges from considering an interaction between the largest values of the generalized Fukui functions fss on both interacting molecules. -
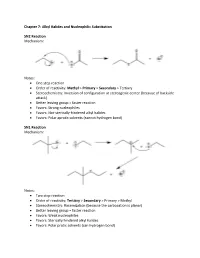
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Reactions of Alkenes Since Bonds Are Stronger Than Bonds, Double Bonds Tend to React to Convert the Double Bond Into Bonds
Reactions of Alkenes Since bonds are stronger than bonds, double bonds tend to react to convert the double bond into bonds This is an addition reaction. (Other types of reaction have been substitution and elimination). Addition reactions are typically exothermic. Electrophilic Addition The bond is localized above and below the C-C bond. The electrons are relatively far away from the nuclei and are therefore loosely bound. An electrophile will attract those electrons, and can pull them away to form a new bond. This leaves one carbon with only 3 bonds and a +ve charge (carbocation). The double bond acts as a nucleophile (attacks the electrophile). Ch08 Reacns of Alkenes (landscape) Page 1 In most cases, the cation produced will react with another nucleophile to produce the final overall electrophilic addition product. Electrophilic addition is probably the most common reaction of alkenes. Consider the electrophilic addition of H-Br to but-2-ene: The alkene abstracts a proton from the HBr, and a carbocation and bromide ion are generated. The bromide ion quickly attacks the cationic center and yields the final product. In the final product, H-Br has been added across the double bond. Ch08 Reacns of Alkenes (landscape) Page 2 Orientation of Addition Consider the addition of H-Br to 2-methylbut-2-ene: There are two possible products arising from the two different ways of adding H-Br across the double bond. But only one is observed. The observed product is the one resulting from the more stable carbocation intermediate. Tertiary carbocations are more stable than secondary.