Catalytic Tuning of Sorption Kinetics of Lightweight Hydrides: a Review of the Materials and Mechanism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Chapter 13.Pptx
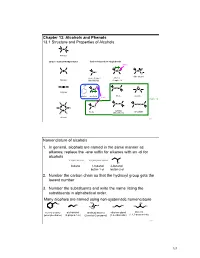
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive. -

Hydrogen Transmutation of Nickel in Glow Discharge
International Journal of Materials Science ISSN 0973-4589 Volume 12, Number 3 (2017), pp. 405-409 © Research India Publications http://www.ripublication.com Hydrogen Transmutation of Nickel in Glow Discharge Vladimir K. Nevolin National Research University of Electronic Technology (MIET), Moscow, Russia. Abstract Background: The possibility of the existence of subatomic hydrogen states was theoretically predicted previously. Objectives: Prove that the transmutation of elements is possible in specially prepared conditions for hydrogen. Methodology: By comparing the mass spectra of deposits on silicon substrates and target electrodes, it is shown that a change in the composition is observed in a magnetron Argon. Results: An increase in the concentration of 62 60 the nickel isotope 28 Ni and a decrease in the isotope concentration 28 Ni are shown. Conclusion: These results confirm the results obtained earlier in the heat generator Rossi, who worked more than a year, found an increase in the 62 isotope 28 Ni due to a decrease in the proportion of other isotopes. Keywords: transmutation, isotopes of nickel, glow discharge, argon, hydrogen INTRODUCION It is considered that the cold transmutation of elements (cold nuclear reactions) has been experimentally demonstrated [1]. On the basis of this phenomenon, energy generators are created in which long-term release of thermal energy in excess of expended energy is observed [2]. From many experimental studies it can be seen that hydrogen, which plays a pivotal role in the reaction zone, may be delivered through a variety of chemical compounds; for example, using lithium aluminium hydride LiAlH4. An analysis of the products of nuclear reactions suggests the possibility of many simultaneous nuclear fusion and decomposition reactions [3]. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

WO 2015/177807 Al 26 November 2015 (26.11.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/177807 Al 26 November 2015 (26.11.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 403/14 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/IN20 15/000066 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 3 February 2015 (03.02.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 1709/MUM/2014 22 May 2014 (22.05.2014) (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: WANBURY LTD. [IN/IN]; BSEL tech park, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, B wing, 10th floor, sector 30A, opp. -

United States Patent Office Patented Aug
3,459,514 United States Patent Office Patented Aug. 5, 1969 2 quired is smaller than the requirements of the prior art. 3,459,514 It is still a further object of the present invention to pro METHOD FOR PREPARING ALKAL vide a process whereby the amount of by-product pro METAL BOROHYDRDES James D. Johnston and Albert P. Giraitis, Baton Rouge, duced is less than that produced by the prior art. Other La., assignors to Ethyl Corporation, New York, N.Y., a objects will become apparent from the ensuing descrip corporation of Virginia tion. No Drawing. Continuation-in-part of applications Ser. No. The above objects are accomplished by the provision 308,691, Sept. 13, 1963, and Ser. No. 322,054, Nov. 7, of a process for producing an alkali metal borohydride 1963. This application Oct. 1, 1964, Ser. No. 400,888 which comprises reacting together an alkali metal hy ret, C. C01b 6/14 O dride, desiccated borax, hydrogen, and silicon at a tem U.S. C. 23-362 6 Claims perature within the range of from about 200 C. to about 900° C. The alkali metal present during the course of the re ABSTRACT OF THE DISCLOSURE action is a member selected from Group I-A of the A method of preparing alkali metal borohydrides com Periodic Chart of the Elements, Fisher Scientific Com prising reacting an alkali metal or alkali metal hydride, pany, 1955. The alkali metals include lithium, sodium, desiccated borax, hydrogen, and silicon, in an inert hy potassium, rubidium, and cesium. Sodium is a preferred drocarbon at about 250° C. -

Lithium Aluminium Hydride.Pdf
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 5.0 Revision Date 03/12/2011 Print Date 09/12/2011 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Aluminium lithium hydride Product Number : 531502 Brand : Aldrich Supplier : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # (For : (314) 776-6555 both supplier and manufacturer) Preparation Information : Sigma-Aldrich Corporation Product Safety - Americas Region 1-800-521-8956 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Water Reactive, Target Organ Effect, Toxic by ingestion, Corrosive Target Organs Kidney, Liver, Central nervous system GHS Classification Substances, which in contact with water, emit flammable gases (Category 1) Acute toxicity, Oral (Category 3) Skin corrosion (Category 1B) Serious eye damage (Category 1) GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H260 In contact with water releases flammable gases which may ignite spontaneously. H301 Toxic if swallowed. H314 Causes severe skin burns and eye damage. Precautionary statement(s) P223 Keep away from any possible contact with water, because of violent reaction and possible flash fire. P231 + P232 Handle under inert gas. Protect from moisture. P280 Wear protective gloves/ protective clothing/ eye protection/ face protection. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P310 Immediately call a POISON CENTER or doctor/ physician. P370 + P378 In case of fire: Use dry sand, dry chemical or alcohol-resistant foam for extinction. P422 Store contents under inert gas. -

Azidocinnamates in Heterocyclic Synthesis
AZIDOCINNAMATES IN HETEROCYCLIC SYNTHESIS A THESIS PRESENTED BY DEIRDRE M.B. HICKEY B.SC. FOR THE DEGREE OF DOCTOR OF PHILOSOPHY j UNIVERSITY OF LONDON Hofmann Laboratory, Department of Chemistry, Imperial College of Science and Technology, London, SW7 2AY. October, 1982. To my parents, Paddy and Thevese 3 with love and gratitude. ACKNOWLEDGEMENTS I thank Professor C.W. Rees for his enthusiastic supervision during the course of this work and my co-supervisor, Dr. C.J. Moody for his constant help and encouragement. I am grateful to Mr. P. Sulsh for technical help, to Mr. D. Neuhaus and Mr. R. Sheppard for Bruker n.m.r. spectra, to Mr. K. Jones for the analytical service, to Mr. J. Bilton, Mrs. Lee, and Mr. N. Davies for the mass spectroscopy service, and to Mr. D. Everitt in the Stores. Miss Moira Shanahan has expertly typed this thesis and I am grateful for her patience and perserverance. I thank Mr. Michael Casey for proof-reading this thesis and for his help and friendship for many years. Dr. Brian Bell I thank for his help and continued support during the production of this thesis. My colleagues on the seventh floor I will warmly remember for their friendship and comradeship. Finally, grateful acknowledgement is made to the Department of Chemistry, Imperial College for a research assistantship. iv ABSTRACT The reactions of various types of nitrenes to give heterocyclic products is reviewed. A series of vinyl azides, mostly ort/zo-substituted azidocinnamates, was prepared and their decomposition reactions studied. The thermal decomposition of ortho-alkyl azidocinnamates gave 2,4- disubstituted indoles, 1,3-disubstituted isoquinolines, 1,3-disubstituted -1,2-dihydroisoquinolines, and enamines, the amounts of each varying with the ortho-group and the conditions used, iodine having a marked effect on the product ratios. -

Sop Pyrophoric 2 12/16/2019
Owner DOC. NO. REV. DATE C.H.O SOP PYROPHORIC 2 12/16/2019 DOC. TITLE SOP FOR PYROPHORIC CHEMICALS Environmental Health & Safety STANDARD OPERATING PROCEDURES (SOP) FOR WORKING WITH PYROPHORIC CHEMICALS AT AMHERST COLLEGE ___________________________________________________________________ General Information Pyrophoric Chemicals are solid, liquid, or gas compounds that, when exposed to air or moisture at or below 54°C (130°F), can spontaneously ignite. Examples of Pyrophoric chemicals used at Amherst College Laboratories include: sodium hydride, zinc powder, and Grignard reagents. See the “Appendix” page below for a full list of Pyrophoric Chemicals. Pyrophoric chemicals are often used as catalysts in chemical reactions or as reducing and deprotonating agents in organic chemistry. Note that Pyrophoric chemicals may also be characterized by other hazards, hence, users of these chemicals may also need to refer to other SOPs that cover other hazards. In addition, each individual chemical’s Safety Data Sheet (SDS) should be consulted before they are used. _____________________________________________________________________________________ Personal Protective Equipment When working with Pyrophoric Chemicals, the following personal protective equipment (PPE) must be worn, at a minimum. Depending on the specific chemical, other forms of protection might be required. Consult the SDS for each chemical before use: Splash goggles Lab coat (Fire resistant lab coat highly recommended) Long pants Close toed shoes Gloves – Nitrile gloves adequate for accidental contact with small quantities. However, the use of fire resistant Nomex/ Leather Pilot’s gloves is highly recommended _____________________________________________________________________________________ Safety Devices All work with Pyrophoric chemicals must be done in a glove box, vacuum manifold, or any enclosed inert environment. If work must be done in a fume hood, ensure that the hood sash is in the lowest feasible position. -

Chem 314 Preorganic Evaluation
Organic Reaction Guide Beauchamp 1 Chem 316 / Beauchamp Reactions Review Sheet Name SN2 Reactions - special features: biomolecular kinetics Rate = kSN2[RX][Nu ], single step concerted reaction, E2 is a competing reaction o o o o relative order of reactivity: CH3X > 1 RX > 2 RX >> 3 RX (based on steric hinderance, no SN2 at 3 RX) allylic & benzylic RX are very reactive, adjacent pi bonds help stabilize transition state and lower TS energy (Ea) o complete substitution at Cα (3 RX) or Cβ (neopentyl pattern) almost completely inhibits SN2 reactions vinyl & phenyl are very unreactive, bonds are stronger and poor backside approach leaving group ability: OTs = I > Br > Cl in neutral or basic conditions (just like E2, SN1 adn E1), and neutral molecule leaving groups are good from protonated, cationic intermediates in acid conditions, + + + + -OH2 , -ORH , -OR2 , -NR3 , etc. we will consider all anions, ammonia, amines, thiols and sulfides to be strong nucleophiles (favors SN2 and E2 reactions) in our course some electron pair donors are mainly nucleophiles (sulfur, azide, cyanide, carboxylates) and - + + - + - some are mainly bases (t-BuO K , Na H2N , Na H ) polar, aprotic solvents work best for SN2 reactions because nucleophiles are relatively unencombered for electron doantion (dimethyl sulofoxide = DMSO, dimethylformamide = DMF, acetonitrile = AN, acetone, etc.) in our course some electron pair donors are mainly nucleophiles (sulfur, azide, cyanide, carboxylates) and we will consider neutral solvent molecules such as water, alcohols and acids to be weak nucleophiles (favors SN1 and E1) stereoselectivity: 100% inversion of configuration from backside atack regioselectivity: reacts at carbon with leaving group, completely unambiguous chemoselectivity: N/A The following list is designed to emphasize SN2 reactions.