The Synthesis of Kinetically Stabilised Heavy Group 13 Hydride Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review of Market for Octane Enhancers
May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado NREL Technical Monitor: K. Ibsen Prepared under Subcontract No. TXE-0-29113-01 National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 NOTICE This report was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or any agency thereof. Available electronically at http://www.doe.gov/bridge Available for a processing fee to U.S. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -

APPENDIX M SUMMARY of MAJOR TYPES of Carfg2 REFINERY
APPENDIX M SUMMARY OF MAJOR TYPES OF CaRFG2 REFINERY MODIFICATIONS Appendix M: Summan/ of Major Types of CaRFG2 Refinery Modifications: Alkylation Units A process unit that combines small-molecule hydrocarbon gases produced in the FCCU with a branched chain hydrocarbon called isobutane, producing a material called alkylate, which is blended into gasoline to raise the octane rating. Alkylate is a high octane, low vapor pressure gasoline blending component that essentially contains no olefins, aromatics, or sulfur. This plant improves the ultimate gasoline-making ability of the FCC plant. Therefore, many California refineries built new or modified existing units to increase alkylate production to blend and to produce greater amounts of CaRFG2. Alkylate is produced by combining C3, C4, and C5 components with isobutane (nC4). The process of alkylation is the reverse of cracking. Olefins (such as butenes and propenes) and isobutane are used as feedstocks and combined to produce alkylate. This process enables refiners to utilize lighter components that otherwise could not be blended into gasoline due to their high vapor pressures. Feed to alkylation unit can include pentanes from light cracked gasoline treaters, isobutanes from butane isomerization unit, and C3/C4 streams from delayed coking units. Isomerization Units - C4/C5/C6 A refinery that has an alkylation plant is not likely to have exactly enough is-butane to match the proplylene and butylene (olefin) feeds. The refiner usually has two choices - buy iso-butane or make it in a butane isomerization (Bl) plant. Isomerization is the rearrangement of straight chain hydrocarbon molecules to form branched chain products or to convert normal paraffins to their isomer. -

Chemical Kinetic Research on HCCI & Diesel Fuels
Lawrence Livermore National Laboratory Chemical Kinetic Research on HCCI & Diesel Fuels William J. Pitz (PI), Charles K. Westbrook, Marco Mehl, M. Lee Davisson Lawrence Livermore National Laboratory May 12, 2009 Project ID # ace_13_pitz DOE National Laboratory Advanced Combustion Engine R&D Merit Review and Peer Evaluation Washington, DC This presentation does not contain any proprietary or confidential information This work performed under the auspices of the U.S. Department of Energy by LLNL-PRES-411416 Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 Overview Timeline Barriers • Ongoing project with yearly • Increases in engine efficiency and direction from DOE decreases in engine emissions are being inhibited by an inadequate ability to simulate in-cylinder combustion and emission formation processes • Chemical kinetic models are critical for Budget improved engine modeling and ultimately for engine design • Funding received in FY08 and expected in FY09: Partners • 800K (DOE) • Interactions/ collaborations: DOE Working Group, National Labs, many universities, FACE Working group. Lawrence Livermore National Laboratory 2 LLNL-PRES-411416LLNL-PRES- 411416 2009 DOE Merit Review Relevance to DOE objectives Support DOE objectives of petroleum-fuel displacement by • Improving engine models so that further improvements in engine efficiency can be made • Developing models for alternative fuels (biodiesel, ethanol, other biofuels, oil-sand derived fuels, Fischer-Tropsch derived fuels) so that they can be better used -
Chapter 13.Pptx
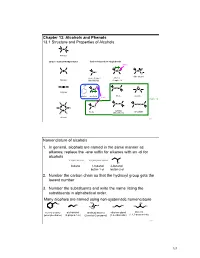
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Exposure Investigation Protocol: the Identification of Air Contaminants Around the Continental Aluminum Plant in New Hudson, Michigan Conducted by ATSDR and MDCH
Exposure Investigation Protocol - Continental Aluminum New Hudson, Lyon Township, Oakland County, Michigan Exposure Investigation Protocol: The Identification of Air Contaminants Around the Continental Aluminum Plant in New Hudson, Michigan Conducted by ATSDR and MDCH MDCH/ATSDR - 2004 Exposure Investigation Protocol - Continental Aluminum New Hudson, Lyon Township, Oakland County, Michigan TABLE OF CONTENTS OBJECTIVE/PURPOSE..................................................................................................... 3 RATIONALE...................................................................................................................... 4 BACKGROUND ................................................................................................................ 5 AGENCY ROLES .............................................................................................................. 6 ESTABLISHING CRITERIA ............................................................................................ 7 “Odor Events”................................................................................................................. 7 Comparison Values......................................................................................................... 7 METHODS ....................................................................................................................... 12 Instantaneous (“Grab”) Air Sampling........................................................................... 12 Continuous Air Monitoring.......................................................................................... -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Synthesis, Characterization, and Reactivity of Chelating Ligand Complexes of Group 13-15 Elements
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 1-1-2019 Synthesis, Characterization, and Reactivity of Chelating Ligand Complexes of Group 13-15 Elements Ala'aeddeen Swidan University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Part of the Chemistry Commons Recommended Citation Swidan, Ala'aeddeen, "Synthesis, Characterization, and Reactivity of Chelating Ligand Complexes of Group 13-15 Elements" (2019). Electronic Theses and Dissertations. 7761. https://scholar.uwindsor.ca/etd/7761 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. Synthesis, Characterization, and Reactivity of Chelating Ligand Complexes of Group 13-15 Elements By Ala’aeddeen Swidan A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the University of Windsor Windsor, Ontario, Canada 2019 © 2019 Ala’aeddeen Swidan Synthesis, Characterization, and Reactivity of Chelating Ligand Complexes of Group 13-15 Elements by Ala’aeddeen Swidan APPROVED BY: ______________________________________________ D. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -

Download The
COORDINATION COMPOUNDS OF ALKYL GALLIUM HYDRIDES by VICTOR GRAHAM WIEBE B.Sc. (Hons.) University of British Columbia 1966 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE In The Department of Chemistry We accept this thesis as conforming to the required standard The University of British Columbia June 1968 In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and Study. I further agree that permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by hits representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. Department of The University of British Columbia Vancouver 8, Canada - ii - Abstract Although the organo hydride derivatives of boron and aluminum are well characterized^little work has been reported on the corresponding gallium systems. The present study was initiated to determine the relative stabilities and reactivity of organo gallium hydride derivatives as compared with the stabilities and reactions of the corresponding compounds of boron and aluminum. Various preparative routes to this new class of gallium compounds have been investigated. These include the use of organo-mercury, organo-lithium and lithium hydride derivatives in reactions with gallium hydride and gallium alkyl compounds and their halogen substituted derivatives: Me3NGaH3 + HgR2 >- Me3NGaH2R + l/2Hg + 1/2H2 Me3NGaH2Cl + LiR y Me3NGaH2R + LiCl Me3NGaR2Cl + LiH • Me3NGaHR2 + LiCl A fourth preparative method involves disproportionation reactions between gallium hydride compounds and organo gallium compounds to yield the mixed organo hydride derivatives. -

The Digallane Molecule, Ga2h6: Experimental Update Giving an Improved Structure and Estimate of the Enthalpy Change for the Reaction Ga2h6(G) 2Gah3(G)
Edinburgh Research Explorer The digallane molecule, Ga2H6: experimental update giving an improved structure and estimate of the enthalpy change for the reaction Ga2H6(g) 2GaH3(g) Citation for published version: Downs, AJ, Greene, TM, Johnsen, E, Pulham, CR, Robertson, HE & Wann, DA 2010, 'The digallane molecule, Ga H : experimental update giving an improved structure and estimate of the enthalpy change for the reaction2 Ga6 H (g) 2GaH (g)', Dalton Transactions, vol. 39, no. 24, pp. 5637-5642. https://doi.org/10.1039/c000694g2 6 3 Digital Object Identifier (DOI): 10.1039/c000694g Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Dalton Transactions General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 PAPER www.rsc.org/dalton | Dalton Transactions The digallane molecule, Ga2H6: experimental update giving an improved structure and estimate of the enthalpy change for the reaction Ga2H6(g) → 2GaH3(g)†‡ Anthony J. Downs,*a Tim M. Greene,a Emma Johnsen,a Colin R.