Chem 314 Preorganic Evaluation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules
ACCOUNT 21 Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules Shunsuke Chiba* Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore Fax +6567911961; E-mail: [email protected] Received 31 May 2012 Organic azides possess diverse chemical reactivities.4 Abstract: In this account, recent advances made on the reactions of several types of organic azides, such as vinyl azides, cyclic 2-azido Owing to their 1,3-dipole character, they undergo [3+2] alcohols, a-azido carbonyl compounds, towards the synthesis of cycloaddition with unsaturated bonds, such as those in nitrogen-containing molecules are described. alkynes and alkenes as well as carbonitriles (Scheme 1, part a).5 Organic azides can also be regarded as nitrene 1 Introduction equivalents (Scheme 1, part b).6 Accordingly, their reac- 2 Chemistry of Vinyl Azides tions with nucleophilic anions, electrophilic cations, and 2.1 Thermal [3+2]-Annulation of Vinyl Azides with 1,3-Dicar- radicals can formally provide the corresponding nitrogen bonyl Compounds 2.2 Manganese(III)-Catalyzed Formal [3+2]-Annulation with anions, cations, and radicals, respectively, forming a new 1,3-Dicarbonyl Compounds bond with the internal azido nitrogen and releasing molec- 2.3 Manganese(III)-Mediated/Catalyzed Formal [3+3]-Annu- ular nitrogen. Moreover, the generation of anions, cations, lation with Cyclopropanols and radicals at the a-position to the azido moiety can re- 2.4 Synthesis of Isoquinolines from a-Aryl-Substituted Vinyl sult in rapid denitrogenation to deliver the corresponding Azides and Internal Alkynes by Rhodium–Copper Bimetal- iminyl species, which can be used in further synthetic lic Cooperation transformations (i.e., carbon–nitrogen bond formation). -
Chapter 13.Pptx
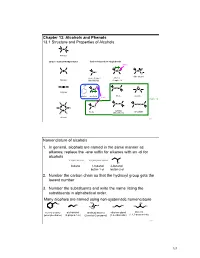
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Safe Handling of Sodium Azide (SAZ)
Safe Handling of Sodium Azide (SAZ) 1,2 Sodium azide (SAZ, CAS# 26628-22-8) is a white crystalline solid [molecular formula of (NaN3)] used in organic synthesis and also as a well-known preservative at low concentrations in molecular biology reagents. Azide chemistry3,4 offers an effective means to synthesize a range of nitrogen-containing compounds with a wide variety of functional groups. But SAZ poses some significant risks. It is highly toxic and can react to form potentially explosive compounds. Azide reagents and intermediates react with some metals5, strong acids, and certain chlorinated solvents6 and this needs to be considered when using SAZ or when developing routes that involve azide-containing intermediates. Health Hazards & Physical Hazards of SAZ Hazard Statements Fatal if swallowed or in contact with skin. Very toxic to aquatic life with long lasting effects. Health Hazards: SAZ is highly toxic when ingested orally or absorbed through the skin. Azides form strong complexes with hemoglobin, and consequently block oxygen transport in the blood. They are more harmful to the heart and the brain than to other organs, because the heart and the brain use a lot of oxygen. Symptoms of exposure include headache, dizziness, nausea and vomiting, rapid breathing and heart rate, and skin burns and blisters (direct skin contact) and in the case of serious overexposure, convulsions and death. It will also react with acids to form hydrazoic acid (HN3). Unlike sodium azide, which is a crystalline solid, hydrazoic acid is a low-boiling, volatile, liquid. Hydrazoic acid is also highly toxic and its volatility makes it more readily inhaled causing lung irritation and potentially bronchitis and lung edema. -

Soluble Guanylate Cyclase B1-Subunit Expression Is Increased in Mononuclear Cells from Patients with Erectile Dysfunction
International Journal of Impotence Research (2006) 18, 432–437 & 2006 Nature Publishing Group All rights reserved 0955-9930/06 $30.00 www.nature.com/ijir ORIGINAL ARTICLE Soluble guanylate cyclase b1-subunit expression is increased in mononuclear cells from patients with erectile dysfunction PJ Mateos-Ca´ceres1, J Garcia-Cardoso2, L Lapuente1, JJ Zamorano-Leo´n1, D Sacrista´n1, TP de Prada1, J Calahorra2, C Macaya1, R Vela-Navarrete2 and AJ Lo´pez-Farre´1 1Cardiovascular Research Unit, Cardiovascular Institute, Hospital Clı´nico San Carlos, Madrid, Spain and 2Urology Department, Fundacio´n Jime´nez Diaz, Madrid, Spain The aim was to determine in circulating mononuclear cells from patients with erectile dysfunction (ED), the level of expression of endothelial nitric oxide synthase (eNOS), soluble guanylate cyclase (sGC) b1-subunit and phosphodiesterase type-V (PDE-V). Peripheral mononuclear cells from nine patients with ED of vascular origin and nine patients with ED of neurological origin were obtained. Fourteen age-matched volunteers with normal erectile function were used as control. Reduction in eNOS protein was observed in the mononuclear cells from patients with ED of vascular origin but not in those from neurological origin. Although sGC b1-subunit expression was increased in mononuclear cells from patients with ED, the sGC activity was reduced. However, only the patients with ED of vascular origin showed an increased expression of PDE-V. This work shows for the first time that, independently of the aetiology of ED, the expression of sGC b1-subunit was increased in circulating mononuclear cells; however, the expression of both eNOS and PDE-V was only modified in the circulating mononuclear cells from patients with ED of vascular origin. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive. -

Cyclohexane Oxidation Continues to Be a Challenge Ulf Schuchardt A,∗, Dilson Cardoso B, Ricardo Sercheli C, Ricardo Pereira A, Rosenira S
Applied Catalysis A: General 211 (2001) 1–17 Review Cyclohexane oxidation continues to be a challenge Ulf Schuchardt a,∗, Dilson Cardoso b, Ricardo Sercheli c, Ricardo Pereira a, Rosenira S. da Cruz d, Mário C. Guerreiro e, Dalmo Mandelli f , Estevam V. Spinacé g, Emerson L. Pires a a Instituto de Qu´ımica, Universidade Estadual de Campinas, P.O. Box 6154, 13083-970 Campinas, SP, Brazil b Depto de Eng. Qu´ımica, Universidade Federal de São Carlos, 13565-905 São Carlos, SP, Brazil c College of Chemistry, University of California, Berkeley, CA 94720, USA d Depto Ciências Exatas e Tecnológicas, Universidade Estadual de Santa Cruz, 45650-000 Ilhéus, BA, Brazil e Universidade Federal de Lavras, Lavras, MG, Brazil f Instituto de Ciências Biológicas e Qu´ımicas, PUC-Campinas, 13020-904 Campinas, SP, Brazil g Sup. Caracterização Qu´ımica, IPEN, 05508-900 São Paulo, SP, Brazil Received 3 October 2000; received in revised form 21 December 2000; accepted 28 December 2000 Abstract Many efforts have been made to develop new catalysts to oxidize cyclohexane under mild conditions. Herein, we review the most interesting systems for this process with different oxidants such as hydrogen peroxide, tert-butyl hydroperoxide and molecular oxygen. Using H2O2, Na-GeX has been shown to be a most stable and active catalyst. Mesoporous TS-1 and Ti-MCM-41 are also stable, but the use of other metals such as Cr, V, Fe and Mo leads to leaching of the metal. Homogeneous systems based on binuclear manganese(IV) complexes have also been shown to be interesting. When t-BuOOH is used, the active systems are those phthalocyanines based on Ru, Co and Cu and polyoxometalates of dinuclear ruthenium and palladium. -

Synthesis and Consecutive Reactions of Α-Azido Ketones: a Review
Molecules 2015, 20, 14699-14745; doi:10.3390/molecules200814699 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Synthesis and Consecutive Reactions of α-Azido Ketones: A Review Sadia Faiz 1,†, Ameer Fawad Zahoor 1,*, Nasir Rasool 1,†, Muhammad Yousaf 1,†, Asim Mansha 1,†, Muhammad Zia-Ul-Haq 2,† and Hawa Z. E. Jaafar 3,* 1 Department of Chemistry, Government College University Faisalabad, Faisalabad-38000, Pakistan, E-Mails: [email protected] (S.F.); [email protected] (N.R.); [email protected] (M.Y.); [email protected] (A.M.) 2 Office of Research, Innovation and Commercialization, Lahore College for Women University, Lahore-54600, Pakistan; E-Mail: [email protected] 3 Department of Crop Science, Faculty of Agriculture, Universiti Putra Malaysia, Serdang-43400, Selangor, Malaysia † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (A.F.Z.); [email protected] (H.Z.E.J.); Tel.: +92-333-6729186 (A.F.Z.); Fax: +92-41-9201032 (A.F.Z.). Academic Editors: Richard A. Bunce, Philippe Belmont and Wim Dehaen Received: 20 April 2015 / Accepted: 3 June 2015 / Published: 13 August 2015 Abstract: This review paper covers the major synthetic approaches attempted towards the synthesis of α-azido ketones, as well as the synthetic applications/consecutive reactions of α-azido ketones. Keywords: α-azido ketones; synthetic applications; heterocycles; click reactions; drugs; azides 1. Introduction α-Azido ketones are very versatile and valuable synthetic intermediates, known for their wide variety of applications, such as in amine, imine, oxazole, pyrazole, triazole, pyrimidine, pyrazine, and amide alkaloid formation, etc. -

Aziridination of Alkenes Promoted by Iron Or Ruthenium Complexes
Aziridination of Alkenes Promoted by Iron or Ruthenium Complexes Caterina Damiano, Daniela Intrieri and Emma Gallo* Department of Chemistry, University of Milan, Via C. Golgi 19, 20133 Milan (Italy). E-mail address: [email protected]. Keywords: Aziridines, Nitrene reagents, Alkenes, Homogenous catalysis, Iron, Ruthenium. Abstract Molecules containing an aziridine functional group are a versatile class of organic synthons due to the presence of a strained three member, which can be easily involved in ring-opening reactions and the aziridine functionality often show interesting pharmaceutical and/or biological behaviours. For these reasons, the scientific community is constantly interested in developing efficient procedures to introduce an aziridine moiety into organic skeletons and the one-pot reaction of an alkene double bond with a nitrene [NR] source is a powerful synthetic strategy. Herein we describe the catalytic activity of iron or ruthenium complexes in promoting the reaction stated above by stressing the potential and limits of each synthetic protocol. 1. Introduction Aziridines, the smallest N-heterocycle compounds, have attracted considerable attention in the last few decades due to their many applications in biological and synthetic chemistry [1]. The aziridine functionality is often responsible for the activity of biologically active species (such as antitumor compounds, antibiotics and enzyme inhibitors) and aziridine containing molecules [2] are also useful building blocks in the synthesis of fine chemicals and pharmaceuticals [3-6]. The striking chemical properties of aziridines are due to the energy associated to the strained three- membered ring [7], which renders them very active and versatile starting materials for the synthesis of several useful molecules such as amines, amino acids, β-lactams, polymers and α-amido ketones [8, 9]. -

United States Patent (19) 11 Patent Number: 6,057,352 Brown Et Al
US006057352A United States Patent (19) 11 Patent Number: 6,057,352 Brown et al. (45) Date of Patent: May 2, 2000 54 FUNGICIDAL CYCLIC AMIDES WO 96/17851 6/1996 WIPO ............................... CO7F 7/08 WO 96/26.191 8/1996 WIPO ... ... CO7D 249/12 75 Inventors: Richard James Brown, Newark, Del.: WO 96/36633 11/1996 WIPO ... ... CO7D 405/04 Deborah Ann Frasier, Martinez, Calif.; WO96/36229 11/1996 WIPO ... ... A01N 43/653 Michael Henry Howard, Jr., WO 97/02255 1/1997 WIPO m CO7D 261/12 Rockland; Gerard Michael Koether, OTHER PUBLICATIONS Bear, both of Del. Zvilichovsky, G., J. Heterocyclic Chem., 24,465–470, 1987. 73 Assignee: E. I. du Pont de Nemours and Zvilichovsky, G. et al., J. Heterocyclic Chem., 25, Company, Wilmington, Del. 1307-1310, 1988. Davis, M. et al., Australian J. Chem., 30(8), 1815-1818, 21 Appl. No.: 08/952,380 1977. 22 PCT Filed: May 8, 1996 Primary Examiner Mukund J. Shah 86 PCT No.: PCT/US96/06534 Assistant Examiner Deepak R. Rao S371 Date: Nov. 13, 1997 57 ABSTRACT S 102(e) Date: Nov. 13, 1997 Compounds of Formula (I), 87 PCT Pub. No.: WO96/36616 (I) PCT Pub. Date: Nov. 21, 1996 1.Y. Nz Related U.S. Application Data x - W 63 Continuation-in-part of application No. 08/442,433, May NY 17, 1995, abandoned. A-N 60 Provisional application No. 60/004,183, Sep. 22, 1995. Ye 51) Int. Cl." ...................... C07D 249/12; CO7D 233/30; AO1N 43/74; AO1N 43/56 and their N-oxides and agriculturally Suitable Salts are 52 U.S. -

Nitric Oxide Activates Guanylate Cyclase and Increases Guanosine 3':5'
Proc. Natl. Acad. Sci. USA Vol. 74, No. 8, pp. 3203-3207, August 1977 Biochemistry Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations (nitro compounds/adenosine 3':5'-cyclic monophosphate/sodium nitroprusside/sodium azide/nitrogen oxides) WILLIAM P. ARNOLD, CHANDRA K. MITTAL, SHOJI KATSUKI, AND FERID MURAD Division of Clinical Pharmacology, Departments of Medicine, Pharmacology, and Anesthesiology, University of Virginia, Charlottesville, Virginia 22903 Communicated by Alfred Gilman, May 16, 1977 ABSTRACT Nitric oxide gas (NO) increased guanylate cy- tigation of this activation. NO activated all crude and partially clase [GTP pyrophosphate-yase (cyclizing), EC 4.6.1.21 activity purified guanylate cyclase preparations examined. It also in- in soluble and particulate preparations from various tissues. The effect was dose-dependent and was observed with all tissue creased cyclic GMP but not adenosine 3':5'-cyclic monophos- preparations examined. The extent of activation was variable phate (cyclic AMP) levels in incubations of minces from various among different tissue preparations and was greatest (19- to rat tissues. 33-fold) with supernatant fractions of homogenates from liver, lung, tracheal smooth muscle, heart, kidney, cerebral cortex, and MATERIALS AND METHODS cerebellum. Smaller effects (5- to 14-fold) were observed with supernatant fractions from skeletal muscle, spleen, intestinal Male Sprague-Dawley rats weighing 150-250 g were decapi- muscle, adrenal, and epididymal fat. Activation was also ob- tated. Tissues were rapidly removed, placed in cold 0.-25 M served with partially purified preparations of guanylate cyclase. sucrose/10 mM Tris-HCl buffer (pH 7.6), and homogenized Activation of rat liver supernatant preparations was augmented in nine volumes of this solution by using a glass homogenizer slightly with reducing agents, decreased with some oxidizing and Teflon pestle at 2-4°.