Regulation of Germinal Center Responses and B-Cell Memory by the Chromatin Modifier MOZ
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -
GM-CSF Receptor-Beta, Human, Recombinant Recombinant Human Granulocyte/Macrophage Colony Stimulating Factor Receptor Beta
GM-CSF Receptor-beta, human, recombinant Recombinant Human Granulocyte/Macrophage Colony Stimulating Factor Receptor beta Instruction Manual Catalog Number C-60430 Synonyms CSF2RB,Colony Stimulating Factor 2 Receptor, Beta, Low-Affinity (Granulocyte-Macrophage), GM-CSF/IL-3/IL-5 Receptor Common Beta Subunit, CDw131, IL3RB, SMDP5, IL5RB, Interleukin 3 Receptor/Granulocyte-Macrophage Colony Stimulating Factor 3 Receptor, Beta (High Affinity), Colony-Stimulating Factor-2 Receptor, Beta, Low-Affinity, GM-CSF/IL-3/IL-5 Receptor Common Beta-Chain, Cytokine Receptor Common Subunit Beta, CD131 Antigen, CD131 Description GM-CSF Receptor-beta, also known as CSF2RB is a member of the type I cytokine receptor family. CSF2RB is a high affinity receptor for interleukin-3, interleukin-5 as well as granulocyte- macrophage colony-stimulating factor. The CSF2RB unique form of receptor assembly applies also to IL-3 and IL-5 receptors, providing a structural basis for understanding their activation mechanism which is essential for the development of therapeutics. Human recombinant GM-CSF Receptor-beta produced in Sf9 cells is a single, glycosylated polypeptide chain of 435 amino acids (17-443 a.a), having a molecular mass of 49.7 kDa (migrates at 40-57 kDa on SDS-PAGE under reducing conditions). CSF2RB is fused to an 8 amino acid His-tag at the C-terminus and purified using proprietary chromatographic techniques. Quantity 10 µg Molecular Mass 49.7 kDa Source Sf9 insect cells Biological-Activity NA Specific Activity NA Formulation Sterile-filtered, clear protein solution (0.5 mg/ml) containing phosphate-buffered saline (pH 7.4) and 10% glycerol. Reconstitution Please Note: Always centrifuge product briefly before opening vial. -

How Relevant Are Bone Marrow-Derived Mast Cells (Bmmcs) As Models for Tissue Mast Cells? a Comparative Transcriptome Analysis of Bmmcs and Peritoneal Mast Cells
cells Article How Relevant Are Bone Marrow-Derived Mast Cells (BMMCs) as Models for Tissue Mast Cells? A Comparative Transcriptome Analysis of BMMCs and Peritoneal Mast Cells 1, 2, 1 1 2,3 Srinivas Akula y , Aida Paivandy y, Zhirong Fu , Michael Thorpe , Gunnar Pejler and Lars Hellman 1,* 1 Department of Cell and Molecular Biology, Uppsala University, The Biomedical Center, Box 596, SE-751 24 Uppsala, Sweden; [email protected] (S.A.); [email protected] (Z.F.); [email protected] (M.T.) 2 Department of Medical Biochemistry and Microbiology, Uppsala University, The Biomedical Center, Box 589, SE-751 23 Uppsala, Sweden; [email protected] (A.P.); [email protected] (G.P.) 3 Department of Anatomy, Physiology and Biochemistry, Swedish University of Agricultural Sciences, Box 7011, SE-75007 Uppsala, Sweden * Correspondence: [email protected]; Tel.: +46-(0)18-471-4532; Fax: +46-(0)18-471-4862 These authors contributed equally to this work. y Received: 29 July 2020; Accepted: 16 September 2020; Published: 17 September 2020 Abstract: Bone marrow-derived mast cells (BMMCs) are often used as a model system for studies of the role of MCs in health and disease. These cells are relatively easy to obtain from total bone marrow cells by culturing under the influence of IL-3 or stem cell factor (SCF). After 3 to 4 weeks in culture, a nearly homogenous cell population of toluidine blue-positive cells are often obtained. However, the question is how relevant equivalents these cells are to normal tissue MCs. By comparing the total transcriptome of purified peritoneal MCs with BMMCs, here we obtained a comparative view of these cells. -
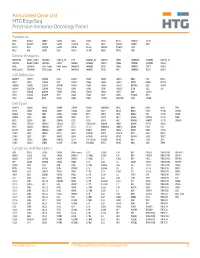
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 KDM5B LMNA NCAPH POC1A SPDL1 BUB1 CDC25C CDKN1B CEP55 ECT2 -

In Situ Observation of Germinal Center Cell Apoptosis During a Secondary Immune Response
J Clin Exp Hematopathol Vol. 46, No. 2, Nov 2006 Original Article In Situ Observation of Germinal Center Cell Apoptosis During a Secondary Immune Response Hito-aki Saitoh, Kunihiko Maeda, and Mitsunori Yamakawa Germinal centers are highly organized anatomic structures essential for the clonal expansion of germinal center (GC) B- cells and associated somatic hypermutation, isotype switching, selection of the high-affinity B-cells (affinity maturation), and elimination of irrelevant or autoreactive clones. The identification of cellular interactions and regulatory mechanisms controlling apoptosis within GCs is essential for a complete understanding of the cellular and molecular dynamics of the GC reaction. We performed a kinetic analysis of the apoptotic activity occurring within GCs of draining lymph nodes of mice immunized with sheep red blood cells (SRBC) after secondary stimulation. The apoptotic activity of GC cells can be divided into three distinct phases : 1) initial phase (within the first days after immunization), 2) reactive phase (from the 5th day to 15th day after secondary immunization), and 3) late phase (after the 15th day). Apoptosis decreased shortly after secondary immunization followed by an increase to peak after an additional 10 days. Finally, apoptosis of GC cells decreased to basal levels. Administration of apoptosis inhibitors decreased the amount of apoptosis during the reactive phase. These results suggest that the reactive phase may be the critical period in which clonal selection and cellular differentiation -

Thymic Lymphoid Follicles in Autoimmune Diseases
Keio J. Med. 20: 57-68. 1971 THYMIC LYMPHOID FOLLICLES IN AUTOIMMUNE DISEASES ‡U. HISTOLOGICAL, HISTOCHEMICAL AND ELECTRON MICROSCOPIC STUDIES NORIKAZU TAMAOKI, SONOKO HABU and TORU KAMEYA Department of Pathology, School of Medicine, Keio University, Tokyo (Received for publication May 8, 1971) Thymic lymphoid follicles have been known to occur frequently in patients of myasthenia gravis, Graves' disease and other autoimmune diseases, and occa sionally in the young subjects dying by accident.1,2,3,4,5 Spontaneous as well as experimental lymphoid follicles have also been described in the thymus of rodents 6,7,8,9 The presense of a thymic lymphoid follicle has been interpreted as an indication of an abnormal immunological reaction, since the thymus is not a site of antibody production under normal conditions. 10 The thymus is an epithelial organ separated from mesenchymal tissue by an epithelial barrier on the capsular surface and around the blood vessels.11,12,13,14 On the other hand, lymphoid follicles are usually found in the peripheral lymphoid tissue composed of mesenchymal reticular cells, such as lymph nodes, spleen andd lymphoid tissue associated with gut. The present study was intended to describe histological, histochemical and electron microscopic characteristics of thymic lymphoid follicles in order to con firm whether they were related to epithelial or mesenchymal elements and to clarify their histogenesis in the human thymus. MATERIALS AND METHODS The materials were the same biospied human thymuses as described in the previous report.5 Serial sections in some cases were made from paraffin blocks and stained alternatively with H & E and silver impregnation. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Germinal Center Formation Motif Signaling in B Cells and Reduced
Altered Regulation of FcγRII on Aged Follicular Dendritic Cells Correlates with Immunoreceptor Tyrosine-Based Inhibition Motif Signaling in B Cells and Reduced This information is current as Germinal Center Formation of October 3, 2021. Yüksel Aydar, Péter Balogh, John G. Tew and Andras K. Szakal J Immunol 2003; 171:5975-5987; ; doi: 10.4049/jimmunol.171.11.5975 Downloaded from http://www.jimmunol.org/content/171/11/5975 References This article cites 47 articles, 17 of which you can access for free at: http://www.jimmunol.org/content/171/11/5975.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 3, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Altered Regulation of Fc␥RII on Aged Follicular Dendritic Cells Correlates with Immunoreceptor Tyrosine-Based Inhibition Motif Signaling in B Cells and Reduced Germinal Center Formation1 Yu¨ksel Aydar,* Pe´ter Balogh,* John G. -

Centers Differentiation Stages in Human Germinal Patterns Reflect
Cutting Edge: Polycomb Gene Expression Patterns Reflect Distinct B Cell Differentiation Stages in Human Germinal Centers This information is current as of September 27, 2021. Frank M. Raaphorst, Folkert J. van Kemenade, Elly Fieret, Karien M. Hamer, David P. E. Satijn, Arie P. Otte and Chris J. L. M. Meijer J Immunol 2000; 164:1-4; ; doi: 10.4049/jimmunol.164.1.1 Downloaded from http://www.jimmunol.org/content/164/1/1 References This article cites 22 articles, 11 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/164/1/1.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2000 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. c Cutting Edge: Polycomb Gene Expression Patterns Reflect Distinct B Cell Differentiation Stages in Human Germinal Centers Frank M. Raaphorst,1* Folkert J. van Kemenade,* Elly Fieret,* Karien M. -

Conserved Transcriptomic Profile Between Mouse and Human Colitis
ARTICLE https://doi.org/10.1038/s41467-019-10769-x OPEN Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification Paulo Czarnewski1, Sara M. Parigi1, Chiara Sorini 1, Oscar E. Diaz 1, Srustidhar Das1, Nicola Gagliani1,2,3 & Eduardo J. Villablanca 1,3 1234567890():,; Clinical manifestations and response to therapies in ulcerative colitis (UC) are hetero- geneous, yet patient classification criteria for tailored therapies are currently lacking. Here, we present an unsupervised molecular classification of UC patients, concordant with response to therapy in independent retrospective cohorts. We show that classical clustering of UC patient tissue transcriptomic data sets does not identify clinically relevant profiles, likely due to associated covariates. To overcome this, we compare cross-sectional human data sets with a newly generated longitudinal transcriptome profile of murine DSS-induced colitis. We show that the majority of colitis risk-associated gene expression peaks during the inflammatory rather than the recovery phase. Moreover, we achieve UC patient clustering into two distinct transcriptomic profiles, differing in neutrophil-related gene activation. Notably, 87% of patients in UC1 cluster are unresponsive to two most widely used biological therapies. These results demonstrate that cross-species comparison enables stratification of patients undistinguishable by other molecular approaches. 1 Immunology and Allergy Unit, Department of Medicine, Solna, Karolinska Institute and University Hospital, 17176 Stockholm, Sweden. 2 Department of Medicine and Department of General, Visceral and Thoracic Surgery, University Medical Center Hamburg-Eppendorf, 20246 Hamburg, Germany. 3These authors jointly supervised this work: Nicola Gagliani, Eduardo J. Villablanca. Correspondence and requests for materials should be addressed to E.J.V. -

Plasma Cells: Finding New Light at the End of B Cell Development Kathryn L
© 2001 Nature Publishing Group http://immunol.nature.com REVIEW Plasma cells: finding new light at the end of B cell development Kathryn L. Calame Plasma cells are cellular factories devoted entire- Upon plasma cell differentiation, there is a marked increase in ly to the manufacture and export of a single prod- steady-state amounts of Ig heavy and light chain mRNA and, when 2 uct: soluble immunoglobulin (Ig). As the final required for IgM and IgA secretion, J chain mRNA . Whether the increase in Ig mRNA is due to increased transcription, increased mediators of a humoral response, plasma cells mRNA stability or, as seems likely, both mechanisms, remains con- play a critical role in adaptive immunity.Although troversial2. There is also an increase in secreted versus membrane intense effort has been devoted to studying the forms of heavy chain mRNA, as determined by differential use of poly(A) sites that may involve the availability of one component of regulation and requirements for early B cell the polyadenylation machinery, cleavage-stimulation factor Cst-643. development, little information has been avail- To accommodate translation and secretion of the abundant Ig able on plasma cells. However, more recent mRNAs, plasma cells have an increased cytoplasmic to nuclear ratio work—including studies on genetically altered and prominent amounts of rough endoplasmic reticulum and secreto- ry vacuoles. mice and data from microarray analyses—has Numerous B cell–specific surface proteins are down-regulated begun to identify the regulatory cascades that upon plasma cell differentiation, including major histocompatibility initiate and maintain the plasma cell phenotype. complex (MHC) class II, B220, CD19, CD21 and CD22. -

Multiple QTL Underlie Milk Phenotypes at the CSF2RB Locus Thomas J
Lopdell et al. Genet Sel Evol (2019) 51:3 https://doi.org/10.1186/s12711-019-0446-x Genetics Selection Evolution RESEARCH ARTICLE Open Access Multiple QTL underlie milk phenotypes at the CSF2RB locus Thomas J. Lopdell1,2*, Kathryn Tiplady1, Christine Couldrey1, Thomas J. J. Johnson1, Michael Keehan1, Stephen R. Davis1, Bevin L. Harris1, Richard J. Spelman1, Russell G. Snell2 and Mathew D. Littlejohn1 Abstract Background: Over many years, artifcial selection has substantially improved milk production by cows. However, the genes that underlie milk production quantitative trait loci (QTL) remain relatively poorly characterised. Here, we investigate a previously reported QTL located at the CSF2RB locus on chromosome 5, for several milk production phe- notypes, to better understand its underlying genetic and molecular causes. Results: Using a population of 29,350 taurine dairy cows, we conducted association analyses for milk yield and composition traits, and identifed highly signifcant QTL for milk yield, milk fat concentration, and milk protein con- centration. Strikingly, protein concentration and milk yield appear to show co-located yet genetically distinct QTL. To attempt to understand the molecular mechanisms that might be mediating these efects, gene expression data were used to investigate eQTL for 11 genes in the broader interval. This analysis highlighted genetic impacts on CSF2RB and NCF4 expression that share similar association signatures to those observed for lactation QTL, strongly implicating one or both of these genes as responsible for these efects. Using the same gene expression dataset representing 357 lactating cows, we also identifed 38 novel RNA editing sites in the 3′ UTR of CSF2RB transcripts.