Vitamin D Modulation of Mitochondrial Oxidative Metabolism and Mtor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

The Suppressive Effects of 1,25-Dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 8-2014 The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration Carolyn Jeanne Ricciardi University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Molecular, Genetic, and Biochemical Nutrition Commons Recommended Citation Ricciardi, Carolyn Jeanne, "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/2876 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Carolyn Jeanne Ricciardi entitled "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Nutrition. -
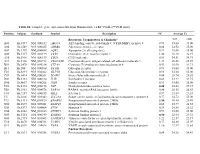
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Nrf1 and Nrf2 Positively and C-Fos and Fra1 Negatively Regulate the Human Antioxidant Response Element-Mediated Expression of NAD(P)H:Quinone Oxidoreductase1 Gene
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 14960–14965, December 1996 Pharmacology Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene RADJENDIRANE VENUGOPAL AND ANIL K. JAISWAL* Department of Pharmacology, Fox Chase Cancer Center, 7701 Burholme Avenue, Philadelphia, PA 19111 Communicated by Y. W. Kan, University of California, San Francisco, CA, August 5, 1996 (received for review February 21, 1996) ABSTRACT Twenty-four base pairs of the human anti- (c-Jun, Jun-D, and Jun-B) form homodimers among them- oxidant response element (hARE) are required for high basal selves and heterodimers with c-Fos and Fra (Fra1 and Fra2) transcription of the NAD(P)H:quinone oxidoreductase1 proteins and bind to the TRE (a single AP1 binding site) and (NQO1) gene and its induction in response to xenobiotics and regulate the expression of target genes (6). Nrf1 and Nrf2 are antioxidants. hARE is a unique cis-element that contains one recently identified NF-E2-related factors that bind to the perfect and one imperfect AP1 element arranged as inverse sequence GCTGAGTCATGATGAGTCA (7, 8). The Nrf1 repeats separated by 3 bp, followed by a ‘‘GC’’ box. We report and Nrf2 contain 742 and 589 amino acids, respectively. Both here that Jun, Fos, Fra, and Nrf nuclear transcription factors these proteins display marked homology to the NF-E2, which bind to the hARE. Overexpression of cDNA derived combi- regulates expression of globin genes in developing erythroid nations of the nuclear proteins Jun and Fos or Jun and Fra1 cells through interaction with upstream AP1-like recognition repressed hARE-mediated chloramphenicol acetyltransferase sites (9). -

Transcription Factors Recognize DNA Shape Without Nucleotide Recognition
bioRxiv preprint doi: https://doi.org/10.1101/143677; this version posted May 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Transcription+factors+recognize+DNA+shape+without+nucleotide+recognition++ Md.$Abul$Hassan$Samee1,$Benoit$G.$Bruneau1,2,$Katherine$S.$Pollard$1,3$ 1:$Gladstone$Institutes,$1650$Owens$St.,$San$Francisco,$CA$94158$ 2:$$Department$of$Pediatrics$and$Cardiovascular$Research$Institute,$University$of$California,$ San$Francisco,$CA$94158$ 3:$Division$of$Bioinformatics,$Institute$for$Human$Genetics,$and$Institute$for$Computational$ Health$Sciences,$University$of$California,$San$Francisco,$CA$94158$ $ + bioRxiv preprint doi: https://doi.org/10.1101/143677; this version posted May 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract+ We$hypothesized$that$transcription$factors$(TFs)$recognize$DNA$shape$without$nucleotide$ sequence$recognition.$Motivating$an$independent$role$for$shape,$many$TF$binding$sites$lack$ a$sequenceZmotif,$DNA$shape$adds$specificity$to$sequenceZmotifs,$and$different$sequences$ can$ encode$ similar$ shapes.$ We$ therefore$ asked$ if$ binding$ sites$ of$ a$ TF$ are$ enriched$ for$ specific$patterns$of$DNA$shapeZfeatures,$e.g.,$helical$twist.$We$developed$ShapeMF,$which$ discovers$ these$ shapeZmotifs$ de%novo%without$ taking$ sequence$ information$ into$ account.$ -

Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks
bioRxiv preprint doi: https://doi.org/10.1101/219667; this version posted November 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Article Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks Weiguang Mao 1,2, Dennis Kostka 1,2,3 and Maria Chikina 1,2* 1 Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh 2 Joint Carnegie Mellon - University of Pittsburgh Ph.D. Program in Computational Biology 3 Department of Developmental Biology, School of Medicine, University of Pittsburgh * Correspondence: [email protected] Academic Editor: name Version November 9, 2017 submitted to Cells 1 Abstract: Background: Gene regulatory sequences play critical roles in ensuring tightly controlled 2 RNA expression patterns that are essential in a large variety of biological processes. Specifically, 3 enhancer sequences drive expression of their target genes, and the availability of genome-wide 4 maps of enhancer-promoter interactions has opened up the possibility to use machine learning 5 approaches to extract and interpret features that define these interactions in different biological 6 contexts. Methods: Inspired by machine translation models we develop an attention-based neural 7 network model, EPIANN, to predict enhancer-promoter interactions based on DNA sequences. Codes 8 and data are available at https://github.com/wgmao/EPIANN. Results: Our approach accurately 9 predicts enhancer-promoter interactions across six cell lines. In addition, our method generates 10 pairwise attention scores at the sequence level, which specify how short regions in the enhancer and 11 promoter pair-up to drive the interaction prediction. -

Influence of DNA Methylation on Transcription Factor Binding
Influence of DNA methylation on transcription factor binding Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Silvia Domcke aus München, Deutschland Basel, 2017 Originaldokument gespeichert auf dem Dokumentenserver der Universität Basel edoc.unibas.ch Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Dirk Schübeler und Dr. François Spitz. Basel, den 18.04.2017 Prof. Dr. Martin Spiess (Dekan) "While Occam's razor is a useful tool in the physical sciences, it can be a very dangerous implement in biology. It is thus very rash to use simplicity and elegance as a guide in biological research. While DNA could be claimed to be both simple and elegant, it must be remembered that DNA almost certainly originated fairly close to the origin of life when things were necessarily simple or they would not have got going. Biologists must constantly keep in mind that what they see was not designed, but rather evolved." ---- Francis Crick in 'What mad Pursuit' (1988) ---- i ii Acknowledgements This thesis would not have been possible without the support and contribution of many people. I would like to thank my PhD advisor Dirk Schübeler for giving me the opportunity to work in his group and for many excellent as well as thought- provoking scientific discussions. I especially appreciate that it was always possible to get critical input and advice while still having a lot of freedom and flexibility. For direct contributions to this study I would like to thank the co-authors of the published manuscript, in particular Anaïs Bardet. -

Supplement. Transcriptional Factors (TF), Protein Name and Their Description Or Function
Supplement. Transcriptional factors (TF), protein name and their description or function. TF Protein name TF description/function ARID3A AT rich interactive domain 3A (BRIGHT-like) This gene encodes a member of the ARID (AT-rich interaction domain) family of DNA binding proteins. ATF4 Activating Transcription Factor 4 Transcriptional activator. Binds the cAMP response element (CRE) (consensus: 5-GTGACGT[AC][AG]-3), a sequence present in many viral and cellular promoters. CTCF CCCTC-Binding Factor Chromatin binding factor that binds to DNA sequence specific sites. Involved in transcriptional regulation by binding to chromatin insulators and preventing interaction between promoter and nearby enhancers and silencers. The protein can bind a histone acetyltransferase (HAT)-containing complex and function as a transcriptional activator or bind a histone deacetylase (HDAC)-containing complex and function as a transcriptional repressor. E2F1-6 E2F transcription factors 1-6 The protein encoded by this gene is a member of the E2F family of transcription factors. The E2F family plays a crucial role in the control of cell cycle and action of tumor suppressor proteins and is also a target of the transforming proteins of small DNA tumor viruses. The E2F proteins contain several evolutionally conserved domains found in most members of the family. These domains include a DNA binding domain, a dimerization domain which determines interaction with the differentiation regulated transcription factor proteins (DP), a transactivation domain enriched in acidic amino acids, and a tumor suppressor protein association domain which is embedded within the transactivation domain. EBF1 Transcription factor COE1 EBF1 has been shown to interact with ZNF423 and CREB binding proteins. -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

Combining Bioinformatics and Experiments to Identify CREB1 As a Key Regulator in Senescent Granulosa Cells
diagnostics Article Combining Bioinformatics and Experiments to Identify CREB1 as a Key Regulator in Senescent Granulosa Cells Pei-Hsuan Lin 1,2, Li-Te Lin 1,3,4, Chia-Jung Li 1,3 , Pei-Gang Kao 1, Hsiao-Wen Tsai 1,3 , San-Nung Chen 1, Zhi-Hong Wen 5 , Peng-Hui Wang 4,6,7,8 and Kuan-Hao Tsui 1,3,4,9,* 1 Department of Obstetrics and Gynecology, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan; [email protected] (P.-H.L.); [email protected] (L.-T.L.); [email protected] (C.-J.L.); [email protected] (P.-G.K.); [email protected] (H.-W.T.); [email protected] (S.-N.C.) 2 Daan Maternal and Children Hospital, Tainan 700, Taiwan 3 Institute of BioPharmaceutical Sciences, National Sun Yat-sen University, Kaohsiung 804, Taiwan 4 Department of Obstetrics and Gynecology, School of Medicine, National Yang-Ming University, Taipei 112, Taiwan; [email protected] 5 Department of Marine Biotechnology and Resources, National Sun Yat-sen University, Kaohsiung 804, Taiwan; [email protected] 6 Department of Obstetrics and Gynecology, Taipei Veterans General Hospital, Taipei 112, Taiwan 7 Department of Medical Research, China Medical University Hospital, Taichung 404, Taiwan 8 Female Cancer Foundation, Taipei 104, Taiwan 9 Department of Pharmacy and Master Program, College of Pharmacy and Health Care, Tajen University, Pingtung County 907, Taiwan * Correspondence: [email protected] Received: 26 April 2020; Accepted: 8 May 2020; Published: 11 May 2020 Abstract: Aging of functional ovaries occurs many years before aging of other organs in the female body. -

ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond
International Journal of Molecular Sciences Review ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond Jun Hamazaki * and Shigeo Murata Laboratory of Protein Metabolism, Graduate School of Pharmaceutical Sciences, the University of Tokyo, Tokyo 1130033, Japan; [email protected] * Correspondence: [email protected] Received: 4 May 2020; Accepted: 20 May 2020; Published: 23 May 2020 Abstract: Protein folding is a substantively error prone process, especially when it occurs in the endoplasmic reticulum (ER). The highly exquisite machinery in the ER controls secretory protein folding, recognizes aberrant folding states, and retrotranslocates permanently misfolded proteins from the ER back to the cytosol; these misfolded proteins are then degraded by the ubiquitin–proteasome system termed as the ER-associated degradation (ERAD). The 26S proteasome is a multisubunit protease complex that recognizes and degrades ubiquitinated proteins in an ATP-dependent manner. The complex structure of the 26S proteasome requires exquisite regulation at the transcription, translation, and molecular assembly levels. Nuclear factor erythroid-derived 2-related factor 1 (Nrf1; NFE2L1), an ER-resident transcription factor, has recently been shown to be responsible for the coordinated expression of all the proteasome subunit genes upon proteasome impairment in mammalian cells. In this review, we summarize the current knowledge regarding the transcriptional regulation of the proteasome, as well as recent findings concerning the regulation of Nrf1 transcription activity in ER homeostasis and metabolic processes. Keywords: proteasome; Nrf1; DDI2; bounce back; proteostasis 1. Introduction Proteins are essential to the vast majority of cellular and organismal functions. Cellular protein levels are defined by the balance among protein synthesis, folding, and degradation, and the failure to accurately coordinate these processes leads to disease [1].