ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

2020 Program Book
PROGRAM BOOK Note that TAGC was cancelled and held online with a different schedule and program. This document serves as a record of the original program designed for the in-person meeting. April 22–26, 2020 Gaylord National Resort & Convention Center Metro Washington, DC TABLE OF CONTENTS About the GSA ........................................................................................................................................................ 3 Conference Organizers ...........................................................................................................................................4 General Information ...............................................................................................................................................7 Mobile App ....................................................................................................................................................7 Registration, Badges, and Pre-ordered T-shirts .............................................................................................7 Oral Presenters: Speaker Ready Room - Camellia 4.......................................................................................7 Poster Sessions and Exhibits - Prince George’s Exhibition Hall ......................................................................7 GSA Central - Booth 520 ................................................................................................................................8 Internet Access ..............................................................................................................................................8 -

2021 Western Medical Research Conference
Abstracts J Investig Med: first published as 10.1136/jim-2021-WRMC on 21 December 2020. Downloaded from Genetics I Purpose of Study Genomic sequencing has identified a growing number of genes associated with developmental brain disorders Concurrent session and revealed the overlapping genetic architecture of autism spectrum disorder (ASD) and intellectual disability (ID). Chil- 8:10 AM dren with ASD are often identified first by psychologists or neurologists and the extent of genetic testing or genetics refer- Friday, January 29, 2021 ral is variable. Applying clinical whole genome sequencing (cWGS) early in the diagnostic process has the potential for timely molecular diagnosis and to circumvent the diagnostic 1 PROSPECTIVE STUDY OF EPILEPSY IN NGLY1 odyssey. Here we report a pilot study of cWGS in a clinical DEFICIENCY cohort of young children with ASD. RJ Levy*, CH Frater, WB Galentine, MR Ruzhnikov. Stanford University School of Medicine, Methods Used Children with ASD and cognitive delays/ID Stanford, CA were referred by neurologists or psychologists at a regional healthcare organization. Medical records were used to classify 10.1136/jim-2021-WRMC.1 probands as 1) ASD/ID or 2) complex ASD (defined as 1 or more major malformations, abnormal head circumference, or Purpose of Study To refine the electroclinical phenotype of dysmorphic features). cWGS was performed using either epilepsy in NGLY1 deficiency via prospective clinical and elec- parent-child trio (n=16) or parent-child-affected sibling (multi- troencephalogram (EEG) findings in an international cohort. plex families; n=3). Variants were classified according to Methods Used We performed prospective phenotyping of 28 ACMG guidelines. -

Representation for Discovery of Protein Motifs
From: ISMB-93 Proceedings. Copyright © 1993, AAAI (www.aaai.org). All rights reserved. Representation for Discovery of Protein Motifs Darrell ConklinT, Suzanne Fortier~, Janice Glasgowt Departments~t of Computing and Information Sciencet and Chemistry Queen’s University Kingston, Ontario, Canada K7L 3N6 [email protected] Abstract pattern of amino acid residues. Protein motifs can be roughly classified into four categories. Sequence There are several dimensions and levels of com- motifs are linear strings of residues with an implicit plexity in which information on protein mo- topological ordering. Sequence-structure motifs are tifs may be available. For example, one- sequence motifs with secondary structure identifiers at- dimensional sequence motifs may be associated tached to one or more residues in the motif. Structure with secondary structure identifiers. Alterna- motifs are 3D structural objects, described by posi- tively, three-dimensional information on polypep- tions of residue objects in 3D Euclidian space. Apart tide segments may be used to induce prototypical from a topological linear ordering on the residues, three-dimensional structure templates. This pa- structure motifs are free of sequence information. Fi- per surveys various representations encountered nally, structure-sequence motifs are combined 1D- in the protein motif discovery literature. Many 3D structures that associate sequence information with of the representations are based on incompatible a structure motif. Figure 1 illustrates these four types semantics, making difficult the comparison and of protein motifs, along with somefurther subclassifi- combination of previous results. To make better cations which are elaborated upon later in the paper. use of machine learning techniques and to pro- The first three motif types are discussed by Thornton vide for an integrated knowledge representation and Gardner (1989); the structure-sequence motif will framework, a general representation language -- be presented in this paper. -

Human Small Maf Proteins Form Heterodimers with CNC Family Transcription Factors and Recognize the NF-E2 Motif
Oncogene (1997) 14, 1901 ± 1910 1997 Stockton Press All rights reserved 0950 ± 9232/97 $12.00 Human small Maf proteins form heterodimers with CNC family transcription factors and recognize the NF-E2 motif Tsutomu Toki1, Jugou Itoh2, Jun'ichi Kitazawa1, Koji Arai1, Koki Hatakeyama3, Jun-itsu Akasaka4, Kazuhiko Igarashi5, Nobuo Nomura6, Masaru Yokoyama1, Masayuki Yamamoto5 and Etsuro Ito1 1Department of Pediatrics, 2Medicine, School of Medicine; 3Department of Biology, Faculty of Sciences, Hirosaki University, Hirosaki 036; 4Department of Biochemistry, Tohoku University School of Medicine, Sendai 980-77; 5Center for TARA and Institute of Basic Medical Sciences, University of Tsukuba, Tsukuba 305; 6Kazusa DNA Institute, Kisarazu 292, Japan The transcription factor NF-E2, a heterodimeric protein Talbot et al., 1990; Talbot and Grosveld, 1991; complex composed of p45 and small Maf family Kotkow and Orkin, 1995). Recent analyses demon- proteins, is considered crucial for the regulation of strated that NF-E2 is composed of two subunits erythroid gene expression and platelet formation. To (Andrews et al., 1993a,b; Igarashi et al., 1994). The facilitate the characterization of NF-E2 functions in large p45 subunit belongs to a family of basic leucine- human cells, we isolated cDNAs encoding two members zipper (bZip) proteins that is closely related to the of the small Maf family, MafK and MafG. The human Drosophila Cap`n'colar (the CNC family) factor mafK and mafG genes encode proteins of 156 and 162 (Mohler et al., 1991). It cannot bind to the NF-E2 amino acid residues, respectively, whose deduced amino sequence as a homodimer, but does do after forming acid sequences show approximately 95% identity to their heterodimers with chicken small Maf family proteins, respective chicken counterparts. -

Distinct Roles of Jun : Fos and Jun : ATF Dimers in Oncogenesis
Oncogene (2001) 20, 2453 ± 2464 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis Hans van Dam*,1 and Marc Castellazzi2 1Department of Molecular Cell Biology, Leiden University Medical Center, Sylvius Laboratories, PO Box 9503, 2300 RA Leiden, The Netherlands; 2Unite de Virologie Humaine, Institut National de la Sante et de la Recherche MeÂdicale (INSERM-U412), Ecole Normale SupeÂrieure, 46 alleÂe d'Italie, 69364 Lyon Cedex 07, France Jun : Fos and Jun : ATF complexes represent two classes dimers with emphasis on their roles in oncogenic of AP-1 dimers that (1) preferentially bind to either transformation in avian model systems. Previous heptameric or octameric AP-1 binding sites, and (2) are reviews on AP-1 and cell transformation include dierently regulated by cellular signaling pathways and references: (Angel and Karin, 1991; Wisdom, 1999; oncogene products. To discriminate between the func- Vogt, 1994; Karin et al., 1997; van Dam and van der tions of Jun : Fos, Jun: ATF and Jun : Jun, mutants were Eb, 1994; Hagmeyer et al., 1995). developed that restrict the ability of Jun to dimerize either to itself, or to Fos(-like) or ATF(-like) partners. Introduction of these mutants in chicken embryo Jun : Fos and Jun : ATF transcription factors: dimeric ®broblasts shows that Jun : Fra2 and Jun : ATF2 dimers complexes with variable composition and activities play distinct, complementary roles in in vitro oncogenesis by inducing either anchorage independence or growth AP-1 sub-units: members of the bZip protein family factor independence, respectively. -

The Suppressive Effects of 1,25-Dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 8-2014 The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration Carolyn Jeanne Ricciardi University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Molecular, Genetic, and Biochemical Nutrition Commons Recommended Citation Ricciardi, Carolyn Jeanne, "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/2876 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Carolyn Jeanne Ricciardi entitled "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Nutrition. -

Maff Is an Antiviral Host Factor That Suppresses Transcription from Hepatitis B Virus Core Promoter
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.29.227793; this version posted March 24, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title 2 MafF is an antiviral host factor that suppresses transcription from Hepatitis B Virus 3 core promoter 4 Running title 5 MafF restricts viral replication 6 Authors 7 Marwa K. Ibrahim1,2, Tawfeek H. Abdelhafez1,2, Junko S. Takeuchi1,3,4, Kosho Wakae1, 8 Masaya Sugiyama5, Masataka Tsuge6, Masahiko Ito7, Koichi Watashi1, Mohamed El Kassas8, 9 Takanobu Kato1, Asako Murayama1, Tetsuro Suzuki7, Kazuaki Chayama9, Kunitada 10 Shimotohno10, Masamichi Muramatsu1#, Hussein H. Aly1#, Takaji Wakita1 11 Affiliation 12 1Department of Virology II, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku-ku, 13 Tokyo 162-8640, Japan 14 2Department of Microbial Biotechnology, Division of Genetic Engineering and Biotechnology 15 Research, National Research Centre, 33 EL Bohouth St. (formerly El Tahrir St.), Dokki, Giza, P. O. 16 12622, Egypt 17 3Center for Clinical Sciences, National Center for Global Health and Medicine, 1-21-1 18 Toyama Shinjuku-ku, Tokyo 162-8655, Japan 19 4Organization for the Strategic Coordination of Research and Intellectual Properties, Meiji 20 University, 1-1-1, Higashi-Mita, Tama-ku, Kawasaki, Kanagawa 214-8571, Japan 21 5Genome Medical Sciences Project, National Center for Global Health and Medicine, 1-7-1 Kohnodai, 22 Ichikawa, Chiba, 272-8516, Japan 23 6Department of Gastroenterology and Metabolism, Graduate School of Biomedical and Health 24 Science, Hiroshima University, 1-2-3 Kasumi, Minami-ku, Hiroshima, 734-8551, Japan 25 7Department of Virology and Parasitology, Hamamatsu University School of Medicine, 1-20-1 26 Handayama, Higashi-ku, Hamamatsu, Shizuoka, 431-3192, Japan 27 8Endemic Medicine Department, Faculty of Medicine, Helwan University, Cairo, P. -

A Drosophila Natural Variation Screen Identifies NKCC1 As a Substrate of NGLY1 Deglycosylation and a Modifier of NGLY1 Deficienc
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.13.039651; this version posted April 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Title 2 A Drosophila natural variation screen identifies NKCC1 as a substrate of NGLY1 deglycosylation 3 and a modifier of NGLY1 deficiency 4 Authors 5 Dana M. Talsness1, Katie G. Owings1, Emily Coelho1, Gaelle Mercenne2, John M. Pleinis2, Aamir 6 R. Zuberi3, Raghavendran Partha4, Nathan L. Clark1, Cathleen M. Lutz3, Aylin R. Rodan2,5, 7 Clement Y. Chow1* 8 The first two authors contributed equally to this study. 1Department of Human Genetics, University 9 of Utah School of Medicine, Salt Lake City, UT; 2Department of Internal Medicine, Division of 10 Nephrology and Hypertension, and Molecular Medicine Program, University of Utah, Salt Lake 11 City, UT; 3Genetic Resource Science, The Jackson Laboratory, Bar Harbor, ME.; 4Department of 12 Computational and Systems Biology, University of Pittsburgh, Pittsburgh, PA; 5Medical Service, 13 Veterans Affairs Salt Lake City Health Care System, Salt Lake City, UT; *For correspondence: 14 [email protected] 15 Abstract 16 N-Glycanase 1 (NGLY1) is a cytoplasmic deglycosylating enzyme. Loss-of-function mutations in 17 the NGLY1 gene cause NGLY1 deficiency, which is characterized by developmental delay, 18 seizures, and a lack of sweat and tears. To model the phenotypic variability observed among 19 patients, we crossed a Drosophila model of NGLY1 deficiency onto a panel of genetically diverse 20 strains. -
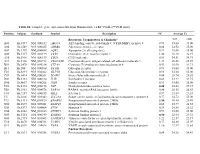
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90