Maff Is an Antiviral Host Factor That Suppresses Transcription from Hepatitis B Virus Core Promoter
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

Human Small Maf Proteins Form Heterodimers with CNC Family Transcription Factors and Recognize the NF-E2 Motif
Oncogene (1997) 14, 1901 ± 1910 1997 Stockton Press All rights reserved 0950 ± 9232/97 $12.00 Human small Maf proteins form heterodimers with CNC family transcription factors and recognize the NF-E2 motif Tsutomu Toki1, Jugou Itoh2, Jun'ichi Kitazawa1, Koji Arai1, Koki Hatakeyama3, Jun-itsu Akasaka4, Kazuhiko Igarashi5, Nobuo Nomura6, Masaru Yokoyama1, Masayuki Yamamoto5 and Etsuro Ito1 1Department of Pediatrics, 2Medicine, School of Medicine; 3Department of Biology, Faculty of Sciences, Hirosaki University, Hirosaki 036; 4Department of Biochemistry, Tohoku University School of Medicine, Sendai 980-77; 5Center for TARA and Institute of Basic Medical Sciences, University of Tsukuba, Tsukuba 305; 6Kazusa DNA Institute, Kisarazu 292, Japan The transcription factor NF-E2, a heterodimeric protein Talbot et al., 1990; Talbot and Grosveld, 1991; complex composed of p45 and small Maf family Kotkow and Orkin, 1995). Recent analyses demon- proteins, is considered crucial for the regulation of strated that NF-E2 is composed of two subunits erythroid gene expression and platelet formation. To (Andrews et al., 1993a,b; Igarashi et al., 1994). The facilitate the characterization of NF-E2 functions in large p45 subunit belongs to a family of basic leucine- human cells, we isolated cDNAs encoding two members zipper (bZip) proteins that is closely related to the of the small Maf family, MafK and MafG. The human Drosophila Cap`n'colar (the CNC family) factor mafK and mafG genes encode proteins of 156 and 162 (Mohler et al., 1991). It cannot bind to the NF-E2 amino acid residues, respectively, whose deduced amino sequence as a homodimer, but does do after forming acid sequences show approximately 95% identity to their heterodimers with chicken small Maf family proteins, respective chicken counterparts. -

Distinct Roles of Jun : Fos and Jun : ATF Dimers in Oncogenesis
Oncogene (2001) 20, 2453 ± 2464 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis Hans van Dam*,1 and Marc Castellazzi2 1Department of Molecular Cell Biology, Leiden University Medical Center, Sylvius Laboratories, PO Box 9503, 2300 RA Leiden, The Netherlands; 2Unite de Virologie Humaine, Institut National de la Sante et de la Recherche MeÂdicale (INSERM-U412), Ecole Normale SupeÂrieure, 46 alleÂe d'Italie, 69364 Lyon Cedex 07, France Jun : Fos and Jun : ATF complexes represent two classes dimers with emphasis on their roles in oncogenic of AP-1 dimers that (1) preferentially bind to either transformation in avian model systems. Previous heptameric or octameric AP-1 binding sites, and (2) are reviews on AP-1 and cell transformation include dierently regulated by cellular signaling pathways and references: (Angel and Karin, 1991; Wisdom, 1999; oncogene products. To discriminate between the func- Vogt, 1994; Karin et al., 1997; van Dam and van der tions of Jun : Fos, Jun: ATF and Jun : Jun, mutants were Eb, 1994; Hagmeyer et al., 1995). developed that restrict the ability of Jun to dimerize either to itself, or to Fos(-like) or ATF(-like) partners. Introduction of these mutants in chicken embryo Jun : Fos and Jun : ATF transcription factors: dimeric ®broblasts shows that Jun : Fra2 and Jun : ATF2 dimers complexes with variable composition and activities play distinct, complementary roles in in vitro oncogenesis by inducing either anchorage independence or growth AP-1 sub-units: members of the bZip protein family factor independence, respectively. -

The Expression of Genes Contributing to Pancreatic Adenocarcinoma Progression Is Influenced by the Respective Environment – Sagini Et Al
The expression of genes contributing to pancreatic adenocarcinoma progression is influenced by the respective environment – Sagini et al Supplementary Figure 1: Target genes regulated by TGM2. Figure represents 24 genes regulated by TGM2, which were obtained from Ingenuity Pathway Analysis. As indicated, 9 genes (marked red) are down-regulated by TGM2. On the contrary, 15 genes (marked red) are up-regulated by TGM2. Supplementary Table 1: Functional annotations of genes from Suit2-007 cells growing in pancreatic environment Categoriesa Diseases or p-Valuec Predicted Activation Number of genesf Functions activationd Z-scoree Annotationb Cell movement Cell movement 1,56E-11 increased 2,199 LAMB3, CEACAM6, CCL20, AGR2, MUC1, CXCL1, LAMA3, LCN2, COL17A1, CXCL8, AIF1, MMP7, CEMIP, JUP, SOD2, S100A4, PDGFA, NDRG1, SGK1, IGFBP3, DDR1, IL1A, CDKN1A, NREP, SEMA3E SERPINA3, SDC4, ALPP, CX3CL1, NFKBIA, ANXA3, CDH1, CDCP1, CRYAB, TUBB2B, FOXQ1, SLPI, F3, GRINA, ITGA2, ARPIN/C15orf38- AP3S2, SPTLC1, IL10, TSC22D3, LAMC2, TCAF1, CDH3, MX1, LEP, ZC3H12A, PMP22, IL32, FAM83H, EFNA1, PATJ, CEBPB, SERPINA5, PTK6, EPHB6, JUND, TNFSF14, ERBB3, TNFRSF25, FCAR, CXCL16, HLA-A, CEACAM1, FAT1, AHR, CSF2RA, CLDN7, MAPK13, FERMT1, TCAF2, MST1R, CD99, PTP4A2, PHLDA1, DEFB1, RHOB, TNFSF15, CD44, CSF2, SERPINB5, TGM2, SRC, ITGA6, TNC, HNRNPA2B1, RHOD, SKI, KISS1, TACSTD2, GNAI2, CXCL2, NFKB2, TAGLN2, TNF, CD74, PTPRK, STAT3, ARHGAP21, VEGFA, MYH9, SAA1, F11R, PDCD4, IQGAP1, DCN, MAPK8IP3, STC1, ADAM15, LTBP2, HOOK1, CST3, EPHA1, TIMP2, LPAR2, CORO1A, CLDN3, MYO1C, -

Nfe2l1) As a Hypoxia- Inducible Factor and Its Oxygen-Dependent Regulation in Vitro and in Vivo
IDENTIFICATION OF NRFl (NFE2L1) AS A HYPOXIA- INDUCIBLE FACTOR AND ITS OXYGEN-DEPENDENT REGULATION IN VITRO AND IN VIVO by NIKOLAI L. CHEPELEV B. Sc. H. Carleton University, 2006 A thesis submitted to the Faculty of Graduate and Postdoctoral Affairs in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology Department of Biology Ottawa-Carleton Institute of Biology Carleton University Ottawa, Ontario, Canada September 2011 © 2011 Nikolai Chepelev Library and Archives Bibliotheque et 1*1 Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington OttawaONK1A0N4 OttawaONK1A0N4 Canada Canada Your file Votre rdterence ISBN: 978-0-494-83249-3 Our file Notre reference ISBN: 978-0-494-83249-3 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library and permettant a la Bibliotheque et Archives Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par I'lnternet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans le loan, distribute and sell theses monde, a des fins commerciales ou autres, sur worldwide, for commercial or non support microforme, papier, electronique et/ou commercial purposes, in microform, autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in this et des droits moraux qui protege cette these. Ni thesis. Neither the thesis nor la these ni des extraits substantiels de celle-ci substantial extracts from it may be ne doivent etre imprimes ou autrement printed or otherwise reproduced reproduits sans son autorisation. -

Genome Sequencing of Xanthomonas Vasicola Pathovar Vasculorum Reveals Variation in Plasmids and Genes Encoding Lipopolysaccharid
Pathogens 2014, 3, 211-237; doi:10.3390/pathogens3010211 OPEN ACCESS pathogens ISSN 2076-0817 www.mdpi.com/journal/pathogens Article Genome Sequencing of Xanthomonas vasicola Pathovar vasculorum Reveals Variation in Plasmids and Genes Encoding Lipopolysaccharide Synthesis, Type-IV Pilus and Type-III Secretion Effectors Arthur Wasukira 1,2, Max Coulter 1, Noorah Al-Sowayeh 1, Richard Thwaites 3, Konrad Paszkiewicz 1, Jerome Kubiriba 2, Julian Smith 3, Murray Grant 1 and David J. Studholme 1,* 1 Biosciences, University of Exeter, Geoffrey Pope Building, Stocker Road, Exeter EX4 4QD, UK; E-Mails: [email protected] (A.W.); [email protected] (M.C.); [email protected] (N.A.-S.); [email protected] (K.P.); [email protected] (M.G.) 2 National Crops Resources Research Institute (NaCRRI), Kampala 7084, Uganda; E-Mail: [email protected] 3 The Food and Environment Research Agency, Sand Hutton, York YO41 1LZ, UK; E-Mails: [email protected] (R.T.); [email protected] (J.S.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +44-(0)-1392-72-4678. Received: 13 December 2013; in revised form: 10 February 2014 / Accepted: 3 March 2014 / Published: 18 March 2014 Abstract: Xanthomonas vasicola pathovar vasculorum (Xvv) is the bacterial agent causing gumming disease in sugarcane. Here, we compare complete genome sequences for five isolates of Xvv originating from sugarcane and one from maize. This identified two distinct types of lipopolysaccharide synthesis gene clusters among Xvv isolates: one is similar to that of Xanthomonas axonopodis pathovar citri (Xac) and is probably the ancestral type, while the other is similar to those of the sugarcane-inhabiting species, Xanthomonas sacchari. -

Engineered Type 1 Regulatory T Cells Designed for Clinical Use Kill Primary
ARTICLE Acute Myeloid Leukemia Engineered type 1 regulatory T cells designed Ferrata Storti Foundation for clinical use kill primary pediatric acute myeloid leukemia cells Brandon Cieniewicz,1* Molly Javier Uyeda,1,2* Ping (Pauline) Chen,1 Ece Canan Sayitoglu,1 Jeffrey Mao-Hwa Liu,1 Grazia Andolfi,3 Katharine Greenthal,1 Alice Bertaina,1,4 Silvia Gregori,3 Rosa Bacchetta,1,4 Norman James Lacayo,1 Alma-Martina Cepika1,4# and Maria Grazia Roncarolo1,2,4# Haematologica 2021 Volume 106(10):2588-2597 1Department of Pediatrics, Division of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 2Stanford Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 3San Raffaele Telethon Institute for Gene Therapy, Milan, Italy and 4Center for Definitive and Curative Medicine, Stanford School of Medicine, Stanford, CA, USA *BC and MJU contributed equally as co-first authors #AMC and MGR contributed equally as co-senior authors ABSTRACT ype 1 regulatory (Tr1) T cells induced by enforced expression of interleukin-10 (LV-10) are being developed as a novel treatment for Tchemotherapy-resistant myeloid leukemias. In vivo, LV-10 cells do not cause graft-versus-host disease while mediating graft-versus-leukemia effect against adult acute myeloid leukemia (AML). Since pediatric AML (pAML) and adult AML are different on a genetic and epigenetic level, we investigate herein whether LV-10 cells also efficiently kill pAML cells. We show that the majority of primary pAML are killed by LV-10 cells, with different levels of sensitivity to killing. Transcriptionally, pAML sensitive to LV-10 killing expressed a myeloid maturation signature. -
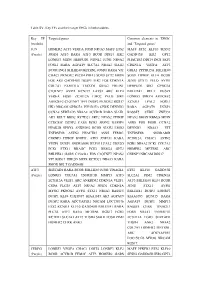
Targeted Genes Common Elements In
Table SV. Key TFs and their target DEGs in hub modules. Key TF Targeted genes Common elements in ‘DEGs’ (module) and ‘Targeted genes’ JUN HOMER2 ATF3 VEGFA FOSB NR4A3 MAFF ETS2 MAFF ETS2 KLF10 SESN2 (Purple) JOSD1 ATF3 RARA ATF3 BCOR DDIT4 IER2 GADD45B IER2 GPT2 LONRF3 MIDN HERPUD1 NDNL2 JUNB NR4A2 PHACTR3 DDIT4 ING1 SKP2 FOSL1 RARA AGPAT9 SLC7A1 NR4A2 SIAH2 CDKN1A VEGFA ATF3 BCOR ING1 BHLHE40 METRNL JOSD1 RARA VIT GRIA2 PPP1R15A BHLHE40 CHAC1 PKNOX2 PLCB4 PHF13 SOX9 SYT2 MIDN SOX9 ITPRIP KLF4 BCOR FOS AK5 GADD45B DUSP1 IER2 FOS CDKN1A JUNB STX11 PELO AVPI1 COL7A1 FAM131A TBCCD1 GRIA2 ERLIN1 HERPUD1 SIK1 GPRC5A C1QTNF7 AVPI1 KCNJ15 LATS2 ARC KLF4 BHLHE41 RELT DUSP2 VEGFA FOSB ZC3H12A LMO2 PELO SIK1 LONRF3 SPRY4 ARHGEF2 ARHGEF2 C1QTNF7 TFPI DUSP2 PKNOX2 RGS17 KCNJ15 LPAL2 FOSL1 HK1 NRCAM GPRC5A PPP1R15A CERK DENND3 RARA AGPAT9 DUSP1 CCNA2 SERTAD3 NR4A2 ACVR1B RARA SIAH1 RASSF5 CERK ZNF331 AK5 RELT BRD2 KCTD21 SKP2 NPAS2 ITPRIP NPAS2 SMOX RBM24 MIDN CCDC85C DUSP2 CARS EGR1 SESN2 RASSF5 ASNS FOS FOSB CCNA2 PIP4K2B SPRY4 ANKRD52 BCOR SIAH2 LMO2 DENND3 NR4A3 VIT TNFRSF1B ASTN2 PHACTR3 ASNS FEM1C TNFRSF1B SNORA80B CSRNP3 ITPRIP BNIP3L ATF3 ZNF331 RARA ZC3H12A CHAC1 ASTN2 VEZF1 DUSP1 SNORA80B KLF10 LPAL2 UBE2O EGR1 NR4A2 PCK1 COL7A1 PCK1 STX11 RRAGC PCK1 RBM24 GPT2 HOMER2 METRNL ARC BHLHE41 GARS C15orf41 FOS C1QTNF7 NPAS2 CSRNP3 NRCAM RGS17 VIT RGS17 UBE2O SOX9 KCTD21 NR4A3 RARA SMOX RELT GADD45B ATF3 SERTAD1 RARA BCOR BHLHE40 JUNB TINAGL1 ETS2 KLF10 GADD45B (Purple) LONRF3 COL7A1 TNFRSF1B MMP13 ATF3 SLC2A1 PIM2 CDKN1A ZC3H12A VEZF1 ARC ANKRD52 -

A MAFG-Lncrna Axis Links Systemic Nutrient Abundance to Hepatic Glucose Metabolism
ARTICLE https://doi.org/10.1038/s41467-020-14323-y OPEN A MAFG-lncRNA axis links systemic nutrient abundance to hepatic glucose metabolism Marta Pradas-Juni et al.# Obesity and type 2 diabetes mellitus are global emergencies and long noncoding RNAs (lncRNAs) are regulatory transcripts with elusive functions in metabolism. Here we show that a high fraction of lncRNAs, but not protein-coding mRNAs, are repressed during diet-induced 1234567890():,; obesity (DIO) and refeeding, whilst nutrient deprivation induced lncRNAs in mouse liver. Similarly, lncRNAs are lost in diabetic humans. LncRNA promoter analyses, global cistrome and gain-of-function analyses confirm that increased MAFG signaling during DIO curbs lncRNA expression. Silencing Mafg in mouse hepatocytes and obese mice elicits a fasting-like gene expression profile, improves glucose metabolism, de-represses lncRNAs and impairs mammalian target of rapamycin (mTOR) activation. We find that obesity-repressed LincIRS2 is controlled by MAFG and observe that genetic and RNAi-mediated LincIRS2 loss causes elevated blood glucose, insulin resistance and aberrant glucose output in lean mice. Taken together, we identify a MAFG-lncRNA axis controlling hepatic glucose metabolism in health and metabolic disease. #A full list of authors and their affiliations appears at the end of the paper. NATURE COMMUNICATIONS | (2020) 11:644 | https://doi.org/10.1038/s41467-020-14323-y | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-14323-y ellular and organism-level energy homeostasis and nutri- by MAFG and CRISPR–Cas9-mediated knockout, or antisense- Cent partitioning are instrumental for survival. In higher mediated RNA interference of LincIRS2 causes hyperglycemia, organisms, multi-organ systems evolved to react to shifts insulin resistance, likely caused by alterations in glucogenic in energy supply by storing (anabolic) or metabolizing (catabolic) gene expression in lean mice. -

ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond
International Journal of Molecular Sciences Review ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond Jun Hamazaki * and Shigeo Murata Laboratory of Protein Metabolism, Graduate School of Pharmaceutical Sciences, the University of Tokyo, Tokyo 1130033, Japan; [email protected] * Correspondence: [email protected] Received: 4 May 2020; Accepted: 20 May 2020; Published: 23 May 2020 Abstract: Protein folding is a substantively error prone process, especially when it occurs in the endoplasmic reticulum (ER). The highly exquisite machinery in the ER controls secretory protein folding, recognizes aberrant folding states, and retrotranslocates permanently misfolded proteins from the ER back to the cytosol; these misfolded proteins are then degraded by the ubiquitin–proteasome system termed as the ER-associated degradation (ERAD). The 26S proteasome is a multisubunit protease complex that recognizes and degrades ubiquitinated proteins in an ATP-dependent manner. The complex structure of the 26S proteasome requires exquisite regulation at the transcription, translation, and molecular assembly levels. Nuclear factor erythroid-derived 2-related factor 1 (Nrf1; NFE2L1), an ER-resident transcription factor, has recently been shown to be responsible for the coordinated expression of all the proteasome subunit genes upon proteasome impairment in mammalian cells. In this review, we summarize the current knowledge regarding the transcriptional regulation of the proteasome, as well as recent findings concerning the regulation of Nrf1 transcription activity in ER homeostasis and metabolic processes. Keywords: proteasome; Nrf1; DDI2; bounce back; proteostasis 1. Introduction Proteins are essential to the vast majority of cellular and organismal functions. Cellular protein levels are defined by the balance among protein synthesis, folding, and degradation, and the failure to accurately coordinate these processes leads to disease [1]. -

A Module of Negative Feedback Regulators Defines Growth Factor
ARTICLES A module of negative feedback regulators defines growth factor signaling Ido Amit1,9, Ami Citri1,8,9, Tal Shay2, Yiling Lu3, Menachem Katz1, Fan Zhang3, Gabi Tarcic1, Doris Siwak3, John Lahad3, Jasmine Jacob-Hirsch4, Ninette Amariglio4, Nora Vaisman5, Eran Segal6, Gideon Rechavi4, Uri Alon7, Gordon B Mills3, Eytan Domany2 & Yosef Yarden1 Signaling pathways invoke interplays between forward signaling and feedback to drive robust cellular response. In this study, we address the dynamics of growth factor signaling through profiling of protein phosphorylation and gene expression, demonstrating the presence of a kinetically defined cluster of delayed early genes that function to attenuate the early events of growth factor signaling. Using epidermal growth factor receptor signaling as the major model system and concentrating on regulation of transcription and mRNA stability, we demonstrate that a number of genes within the delayed early gene cluster function as feedback regulators of immediate early genes. Consistent with their role in negative regulation of cell signaling, genes within http://www.nature.com/naturegenetics this cluster are downregulated in diverse tumor types, in correlation with clinical outcome. More generally, our study proposes a mechanistic description of the cellular response to growth factors by defining architectural motifs that underlie the function of signaling networks. Cells respond in a stereotypic and highly reproducible manner to phosphorylation and gene expression, with the aim of uncovering external stimuli. A major focus of current research is elucidation of the mechanisms of transcription-dependent signal attenuation. Conse- mechanisms underlying the regulation of this response. In the past, quently, we can now identify a cluster of coexpressed genes that considerable effort was invested in characterizing ‘forward-signaling’ function in feedback attenuation of growth factor signaling at specific components activated by external stimuli, with successful identifica- nodes within the network (for an overview, see Supplementary Fig. -

Integrative Analysis Identifies Bhlh Transcription Factors As Contributors to Parkinson's Disease Risk Mechanisms
www.nature.com/scientificreports OPEN Integrative analysis identifes bHLH transcription factors as contributors to Parkinson’s disease risk mechanisms Victoria Berge‑Seidl1,2, Lasse Pihlstrøm 1 & Mathias Toft 1,2* Genome‑wide association studies (GWAS) have identifed multiple genetic risk signals for Parkinson’s disease (PD), however translation into underlying biological mechanisms remains scarce. Genomic functional annotations of neurons provide new resources that may be integrated into analyses of GWAS fndings. Altered transcription factor binding plays an important role in human diseases. Insight into transcriptional networks involved in PD risk mechanisms may thus improve our understanding of pathogenesis. We analysed overlap between genome‑wide association signals in PD and open chromatin in neurons across multiple brain regions, fnding a signifcant enrichment in the superior temporal cortex. The involvement of transcriptional networks was explored in neurons of the superior temporal cortex based on the location of candidate transcription factor motifs identifed by two de novo motif discovery methods. Analyses were performed in parallel, both fnding that PD risk variants signifcantly overlap with open chromatin regions harboring motifs of basic Helix‑Loop‑Helix (bHLH) transcription factors. Our fndings show that cortical neurons are likely mediators of genetic risk for PD. The concentration of PD risk variants at sites of open chromatin targeted by members of the bHLH transcription factor family points to an involvement of these transcriptional networks in PD risk mechanisms. Parkinson’s disease (PD) is a progressive neurodegenerative disorder afecting about 1% of the population above 60 years of age1. Te cause of neuronal death is poorly understood, and this is obstructing the path toward more efective treatments.