Science Journals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Detection of Interacting Transcription Factors in Human Tissues Using
Myšičková and Vingron BMC Genomics 2012, 13(Suppl 1):S2 http://www.biomedcentral.com/1471-2164/13/S1/S2 PROCEEDINGS Open Access Detection of interacting transcription factors in human tissues using predicted DNA binding affinity Alena Myšičková*, Martin Vingron From The Tenth Asia Pacific Bioinformatics Conference (APBC 2012) Melbourne, Australia. 17-19 January 2012 Abstract Background: Tissue-specific gene expression is generally regulated by combinatorial interactions among transcription factors (TFs) which bind to the DNA. Despite this known fact, previous discoveries of the mechanism that controls gene expression usually consider only a single TF. Results: We provide a prediction of interacting TFs in 22 human tissues based on their DNA-binding affinity in promoter regions. We analyze all possible pairs of 130 vertebrate TFs from the JASPAR database. First, all human promoter regions are scanned for single TF-DNA binding affinities with TRAP and for each TF a ranked list of all promoters ordered by the binding affinity is created. We then study the similarity of the ranked lists and detect candidates for TF-TF interaction by applying a partial independence test for multiway contingency tables. Our candidates are validated by both known protein-protein interactions (PPIs) and known gene regulation mechanisms in the selected tissue. We find that the known PPIs are significantly enriched in the groups of our predicted TF-TF interactions (2 and 7 times more common than expected by chance). In addition, the predicted interacting TFs for studied tissues (liver, muscle, hematopoietic stem cell) are supported in literature to be active regulators or to be expressed in the corresponding tissue. -

Identifying and Mapping Cell-Type-Specific Chromatin PNAS PLUS Programming of Gene Expression
Identifying and mapping cell-type-specific chromatin PNAS PLUS programming of gene expression Troels T. Marstranda and John D. Storeya,b,1 aLewis-Sigler Institute for Integrative Genomics, and bDepartment of Molecular Biology, Princeton University, Princeton, NJ 08544 Edited by Wing Hung Wong, Stanford University, Stanford, CA, and approved January 2, 2014 (received for review July 2, 2013) A problem of substantial interest is to systematically map variation Relating DHS to gene-expression levels across multiple cell in chromatin structure to gene-expression regulation across con- types is challenging because the DHS represents a continuous ditions, environments, or differentiated cell types. We developed variable along the genome not bound to any specific region, and and applied a quantitative framework for determining the exis- the relationship between DHS and gene expression is largely tence, strength, and type of relationship between high-resolution uncharacterized. To exploit variation across cell types and test chromatin structure in terms of DNaseI hypersensitivity and genome- for cell-type-specific relationships between DHS and gene expres- wide gene-expression levels in 20 diverse human cell types. We sion, the measurement units must be placed on a common scale, show that ∼25% of genes show cell-type-specific expression ex- the continuous DHS measure associated to each gene in a well- plained by alterations in chromatin structure. We find that distal defined manner, and all measurements considered simultaneously. regions of chromatin structure (e.g., ±200 kb) capture more genes Moreover, the chromatin and gene-expression relationship may with this relationship than local regions (e.g., ±2.5 kb), yet the local only manifest in a single cell type, making standard measures of regions show a more pronounced effect. -

REST Mediates Androgen Receptor Actions on Gene Repression And
Published online 24 October 2013 Nucleic Acids Research, 2014, Vol. 42, No. 2 999–1015 doi:10.1093/nar/gkt921 REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer Charlotte Svensson1, Jens Ceder2, Diego Iglesias-Gato1, Yin-Choy Chuan1, See Tong Pang3, Anders Bjartell2, Roxana Merino Martinez4, Laura Bott5, Leszek Helczynski6, David Ulmert2,7, Yuzhuo Wang8, Yuanjie Niu9, Colin Collins8 and Amilcar Flores-Morales1,* 1Novo Nordisk Foundation Center for Protein Research, Faculty of Health Sciences, University of Copenhagen, Downloaded from DK-2200 Copenhagen, Denmark, 2Division of Urological Cancers, Department of Clinical Sciences, Ska˚ ne University Hospital, Lund University, 20502 Malmo¨ , Sweden, 3Department of Urology, Chang Gung Memorial Hospital, Tao-Yuan 33305, Taiwan, R.O.C., 4Department of Epidemiology, Karolinska Institutet, 171 77 Stockholm, Sweden, 5Department of Cell and Molecular Biology, Karolinska Institute, 171 77 Stockholm, Sweden, 6Regional Laboratories Region Ska˚ ne, Clinical Pathology, 205 80 Malmo¨ , Sweden, 7Department of Surgery (Urology), Memorial Sloan-Kettering Cancer Center, New York, NY 100 65, USA, 8Vancouver Prostate http://nar.oxfordjournals.org/ Centre and The Department of Urologic Sciences, University of British Columbia, Vancouver, BC Canada V6H 3Z6 and 9Tianjin Institute of Urology, Tianjin Medical University, Tianjin 300 211, China Received December 19, 2012; Accepted September 20, 2013 ABSTRACT that has previously been implicated in the growth at University of British Columbia on August 12, 2014 The androgen receptor (AR) is a key regulator of NE-like castration-resistant tumors. The of prostate tumorgenesis through actions that are analysis of prostate cancer tissue microarrays not fully understood. We identified the repressor revealed that tumors with reduced expression of element (RE)-1 silencing transcription factor REST have higher probability of early recurrence, (REST) as a mediator of AR actions on gene repres- independently of their Gleason score. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Elucidation of the ELK1 Target Gene Network Reveals a Role in the Coordinate Regulation of Core Components of the Gene Regulation Machinery
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery Joanna Boros,1,5 Ian J. Donaldson,1,5 Amanda O’Donnell,1 Zaneta A. Odrowaz,1 Leo Zeef,1 Mathieu Lupien,2,4 Clifford A. Meyer,3 X. Shirley Liu,3 Myles Brown,2 and Andrew D. Sharrocks1,6 1Faculty of Life Sciences, University of Manchester, Manchester M13 9PT, United Kingdom; 2Division of Molecular and Cellular Oncology, Department of Medical Oncology, Dana-Farber Cancer Institute and Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts 02115, USA; 3Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute and Harvard School of Public Health, Boston, Massachusetts 02115, USA Transcription factors play an important role in orchestrating the activation of specific networks of genes through targeting their proximal promoter and distal enhancer regions. However, it is unclear how the specificity of downstream responses is maintained by individual members of transcription-factor families and, in most cases, what their target repertoire is. We have used ChIP-chip analysis to identify the target genes of the ETS-domain transcription factor ELK1. Two distinct modes of ELK1 target gene selection are identified; the first involves redundant promoter binding with other ETS-domain family members; the second occurs through combinatorial binding with a second transcription factor SRF, which specifies a unique group of target genes. One of the most prominent groups of genes forming the ELK1 target network includes classes involved in core gene expression control, namely, components of the basal transcriptional machinery, the spliceosome and the ribosome. -

SLC45A3-ELK4 Is a Novel and Frequent Erythroblast Transformation–Specific Fusion Transcript in Prostate Cancer
Published OnlineFirst March 17, 2009; DOI: 10.1158/0008-5472.CAN-08-4926 Priority Report SLC45A3-ELK4 Is a Novel and Frequent Erythroblast Transformation–Specific Fusion Transcript in Prostate Cancer David S. Rickman,1 Dorothee Pflueger,1 Benjamin Moss,1 Vanessa E. VanDoren,1 Chen X. Chen,1 Alexandre de la Taille,4,5 Rainer Kuefer,6 Ashutosh K. Tewari,2 Sunita R. Setlur,7 Francesca Demichelis,1,3 and Mark A. Rubin1 Departments of 1Pathology and Laboratory Medicine and 2Urology, and 3Institute for Computational Biomedicine, Weill Cornell Medical College, New York, New York; 4Department of Urology, CHU Mondor and 5Institut National de la Sante et de la Recherche Medicale, Unite´841, Cre´teil,France; 6Department of Urology, University Hospital Ulm, Ulm, Germany; and 7Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts Abstract (2, 3), and a specific expression profile (4, 5). Androgen-regulated ¶ Chromosomal rearrangements account for all erythroblast genes account for the majority of the 5 genomic regulatory transformation–specific (ETS) family member gene fusions promoters elements fused with ETS genes in prostate cancer (6). The promoter of the androgen-regulated transmembrane protease, that have been reported in prostate cancer and have clinical, serine 2 TMPRSS2 diagnostic, and prognostic implications. Androgen-regulated ( ) gene is fused to the coding region of members genes account for the majority of the 5¶ genomic regulatory of the ETS family of transcription factors, most commonly v-ets TMPRSS2-ERG, erythroblastosis virus E26 oncogene homologue (avian; ERG; ref. 7). promoter elements fused with ETS genes. Solute carrier family 45, member 3 SLC45A3 TMPRSS2-ETV1, and SLC45A3-ERG rearrangements account ( ), also referred to as for roughly 90% of ETS fusion prostate cancer. -

The Suppressive Effects of 1,25-Dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 8-2014 The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration Carolyn Jeanne Ricciardi University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Molecular, Genetic, and Biochemical Nutrition Commons Recommended Citation Ricciardi, Carolyn Jeanne, "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/2876 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Carolyn Jeanne Ricciardi entitled "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Nutrition. -
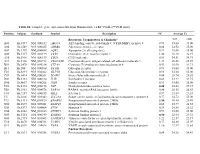
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Nrf1 and Nrf2 Positively and C-Fos and Fra1 Negatively Regulate the Human Antioxidant Response Element-Mediated Expression of NAD(P)H:Quinone Oxidoreductase1 Gene
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 14960–14965, December 1996 Pharmacology Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene RADJENDIRANE VENUGOPAL AND ANIL K. JAISWAL* Department of Pharmacology, Fox Chase Cancer Center, 7701 Burholme Avenue, Philadelphia, PA 19111 Communicated by Y. W. Kan, University of California, San Francisco, CA, August 5, 1996 (received for review February 21, 1996) ABSTRACT Twenty-four base pairs of the human anti- (c-Jun, Jun-D, and Jun-B) form homodimers among them- oxidant response element (hARE) are required for high basal selves and heterodimers with c-Fos and Fra (Fra1 and Fra2) transcription of the NAD(P)H:quinone oxidoreductase1 proteins and bind to the TRE (a single AP1 binding site) and (NQO1) gene and its induction in response to xenobiotics and regulate the expression of target genes (6). Nrf1 and Nrf2 are antioxidants. hARE is a unique cis-element that contains one recently identified NF-E2-related factors that bind to the perfect and one imperfect AP1 element arranged as inverse sequence GCTGAGTCATGATGAGTCA (7, 8). The Nrf1 repeats separated by 3 bp, followed by a ‘‘GC’’ box. We report and Nrf2 contain 742 and 589 amino acids, respectively. Both here that Jun, Fos, Fra, and Nrf nuclear transcription factors these proteins display marked homology to the NF-E2, which bind to the hARE. Overexpression of cDNA derived combi- regulates expression of globin genes in developing erythroid nations of the nuclear proteins Jun and Fos or Jun and Fra1 cells through interaction with upstream AP1-like recognition repressed hARE-mediated chloramphenicol acetyltransferase sites (9). -

Pharmacodynamic Effects of Seliciclib, an Orally Administered
Cancer Therapy: Clinical Pharmacodynamic Effects of Seliciclib, an Orally Administered Cell Cycle Modulator, in Undifferentiated Nasopharyngeal Cancer Wen-Son Hsieh,4 Ross Soo,1Bee-Keow Peh,2 Thomas Loh,4 Difeng Dong,3 Donny Soh,5 Lim-SoonWong,3,5 Simon Green,6 Judy Chiao,6 Chun-Ying Cui,1Yo k e -F o n g L a i , 4 Soo-Chin Lee,1Benjamin Mow,1 Richie Soong,2 Manuel Salto-Tellez,2 and Boon-Cher Goh1, 2 Abstract Purpose: Cell cycle dysregulation resulting in expression of antiapoptotic genes and uncontrolled proliferation is a feature of undifferentiated nasopharyngeal carcinoma. The pharmacodynamic effects of seliciclib, a cyclin-dependent kinase (CDK) inhibitor, were studied in patients with nasopharyngeal carcinoma. Experimental Design: Patients with treatment-naI«ve locally advanced nasopharyngeal carci- noma received seliciclib at 800 mg or 400 mg twice daily on days1 to 3 and 8 to12.Paired tumor samples obtained at baseline and on day 13 were assessed by light microscopy, immunohisto- chemistry, and transcriptionalprofiling using real-time PCR low-density array consisting of apanel of 380 genes related to cell cycle inhibition, apoptosis, signal transduction, and cell proliferation. Results: At 800 mg bd, one patient experienced grade 3 liver toxicity and another had grade 2 vomiting; no significant toxicities were experienced in 13 patients treated at 400 mg bd. Seven of fourteen evaluable patients had clinical evidence of tumor reduction. Some of these responses were associated with increased tumor apoptosis, necrosis, and decreases in plasma EBV DNA posttreatment. Reduced protein expression of Mcl-1, cyclin D1, phosphorylated retinoblastoma protein pRB (T821), and significant transcriptional down-regulation of genes related to cellular proliferation and survival were shown in some patients posttreatment, indicative of cell cycle modulation by seliciclib, more specifically inhibition of cdk2/cyclin E, cdk7/cyclin H, and cdk9/cyclinT.