Australian Public Assessment Report for Odomzo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cyclopamine Inhibition of Sonic Hedgehog Signal Transduction Is
Developmental Biology 224, 440–452 (2000) doi:10.1006/dbio.2000.9775, available online at http://www.idealibrary.com on View metadata, citation and similar papers at core.ac.uk brought to you by CORE Cyclopamine Inhibition of Sonic Hedgehog Signalprovided by Elsevier - Publisher Connector Transduction Is Not Mediated through Effects on Cholesterol Transport John P. Incardona,* William Gaffield,† Yvonne Lange,‡ Adele Cooney,§ Peter G. Pentchev,§ Sharon Liu,¶ John A. Watson,¶ Raj P. Kapur,ሻ and Henk Roelink*,1 *Department of Biological Structure and Center for Developmental Biology and Department of Pathology, University of Washington, Seattle, Washington 98195; †Western Regional Research Center, ARS, United States Department of Agriculture, Albany, California 94710; ‡Department of Pathology, Rush–Presbyterian–St. Luke’s Medical Center, Chicago, Illinois 60612; §Developmental and Metabolic Neurobiology Branch, NINDS, National Institutes of Health, Bethesda, Maryland 20892; and ¶Department of Biochemistry and Biophysics, University of California at San Francisco, San Francisco, California 94143 Cyclopamine is a teratogenic steroidal alkaloid that causes cyclopia by blocking Sonic hedgehog (Shh) signal transduction. We have tested whether this activity of cyclopamine is related to disruption of cellular cholesterol transport and putative secondary effects on the Shh receptor, Patched (Ptc). First, we report that the potent antagonism of Shh signaling by cyclopamine is not a general property of steroidal alkaloids with similar structure. The structural features of steroidal alkaloids previously associated with the induction of holoprosencephaly in whole animals are also associated with inhibition of Shh signaling in vitro. Second, by comparing the effects of cyclopamine on Shh signaling with those of compounds known to block cholesterol transport, we show that the action of cyclopamine cannot be explained by inhibition of intracellular cholesterol transport. -

Investigator Initiated Study IRB-29839 an Open-Label Pilot Study To
Investigator Initiated Study IRB-29839 An open-label pilot study to evaluate the efficacy and safety of a combination treatment of Sonidegib and BKM120 for the treatment of advanced basal cell carcinomas Version 05 September 2016 NCT02303041 DATE: 12Dec2018 1 Coordinating Center Stanford Cancer Center 875 Blake Wilbur Drive Stanford, CA 94305 And 450 Broadway, MC 5334 Redwood City, CA 94603 Protocol Director and Principal Investigator Anne Lynn S Chang, MD, Director of Dermatological Clinical Trials 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] Co-Investigator Anthony Oro, MD PhD 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] Biostatistician Shufeng Li, MS 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] Study Coordinator Ann Moffat 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] 2 Table of Contents 1 Background ................................................................. 7 1.1 Disease Background ..................................................... 7 1.2 Hedgehog Pathway and mechanism of action ............................... 7 1.3 PI3K Pathway and mechanism of action ................................... 9 1.4 Sonidegib Compound Information ............ Error! Bookmark not defined. 1.4.1 Preclinical Studies for Sonidegib ....................................................................11 1.4.2 Muscular system...............................................................................................13 1.4.3 Skeletal system ................................................................................................13 -
FDA-Approved Content Report Section 1
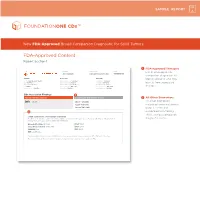
SAMPLE REPORT New FDA-Approved Broad Companion Diagnostic for Solid Tumors FDA-Approved Content Report Section 1 1 FDA-Approved Therapies PATIENT TUMOR TYPE TRF# List of FDA-approved Jane Sample Lung adenocarcinoma TRFXXXXXX companion diagnostics to PATIENT PHYSICIAN SPECIMEN identify patients who may DISEASE Lung adenocarcinoma ORDERING PHYSICIAN Not Given SPECIMEN SITE Not Given NAME Not Given MEDICAL FACILITY Not Given SPECIMEN ID Not Given benefi t from associated DATE OF BIRTH Not Given ADDITIONAL RECIPIENT Not Given SPECIMEN TYPE Not Given SEX Female MEDICAL FACILITY ID Not Given DATE OF COLLECTION Not Given therapies MEDICAL RECORD # Not Given PATHOLOGIST Not Given SPECIMEN RECEIVED Not Given CDx Associated Findings 1 GENOMIC FINDINGS DETECTED FDA-APPROVED THERAPEUTIC OPTIONS 2 All Other Biomarkers EGFR L858R Gilotrif® (Afatinib) All other biomarkers, Iressa® (Gefitinib) including tumor mutational Tarceva® (Erlotinib) burden (TMB) and 2 microsatellite instability (MSI), without companion OTHER ALTERATIONS & BIOMARKERS IDENTIFIED Results reported in this section are not prescriptive or conclusive for labeled use of any specific therapeutic product. See diagnostic claims professional services section for additional information. Microsatellite Status MS-Stable PTCH1 T416S Tumor Mutation Burden 11 Muts/Mb RBM10 Q494* CDKN2A/B loss TP53 R267P EGFR amplification § Refer to appendix for limitation statements related to detection of any copy number alterations, gene rearrangements, MSI or TMB result in this section. Please refer to appendix -

An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib Christina Danial, Kavita Y
Published OnlineFirst November 6, 2015; DOI: 10.1158/1078-0432.CCR-15-1588 Clinical Trial Brief Report Clinical Cancer Research An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib Christina Danial, Kavita Y. Sarin, Anthony E. Oro, and Anne Lynn S. Chang Abstract Purpose: To assess the tumor response to the smoothened sive disease with sonidegib. Three patients experienced stable (SMO) inhibitor, sonidegib (LDE225), in patients with an disease and discontinued sonidegib either due to adverse events advanced basal cell carcinoma (BCC) resistant to treatment with (n ¼ 1) or due to election for surgery (n ¼ 2). The response of one vismodegib (GDC0449). patient was not evaluable. SMO mutations with in vitro data Experimental Design: Nine patients with an advanced suggesting resistance to Hh pathway inhibition were identified BCC that was previously resistant to treatment with vismode- in 5 patients, and none of these patients experienced responses gib were given sonidegib in this investigational, open- while on sonidegib. label study. Tumor response was determined using the Conclusion: Patients with advanced BCCs that were response evaluation criteria in solid tumors. SMO mutations previously resistant to treatment with vismodegib similarly were identified using biopsy samples from the target BCC demonstrated treatment resistance with sonidegib. Patients location. who have developed treatment resistance to an SMO inhibitor Results: The median duration of treatment with sonidegib was may continue to experience tumor progression in response to 6 weeks (range, 3–58 weeks). Five patients experienced progres- other SMO inhibitors. Clin Cancer Res; 1–5. Ó2015 AACR. Introduction Sonidegib (LDE225) is a new SMO inhibitor approved in 2015 by the FDA for locally advanced BCCs. -

Sonidegib for the Treatment of Advanced Basal Cell Carcinoma
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2016 Sonidegib for the treatment of advanced basal cell carcinoma Ramelyte, Egle ; Amann, Valerie C ; Dummer, Reinhard DOI: https://doi.org/10.1080/14656566.2016.1225725 Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-125862 Journal Article Accepted Version Originally published at: Ramelyte, Egle; Amann, Valerie C; Dummer, Reinhard (2016). Sonidegib for the treatment of advanced basal cell carcinoma. Expert Opinion on Pharmacotherapy, 17(14):1963-1968. DOI: https://doi.org/10.1080/14656566.2016.1225725 Sonidegib for the treatment of advanced basal cell carcinoma Egle Ramelyte1,2,MD, Valerie C. Amann1, MD, Reinhard Dummer 1, MD, 1 - Department of Dermatology, University Hospital Zurich, Zurich, Switzerland 2 - Vilnius University, Centre of Dermatovenereology, Vilnius, Lithuania Corresponding author: Reinhard Dummer, MD University Hospital of Zurich Department of Dermatology Gloriastrasse 31 CH-8091, Zürich, Switzerland Phone: 41-44-255-2507 Fax: 41-44-255-8988 E-mail: [email protected] 1 Abstract Introduction. Advanced and metastatic basal cell carcinomas (BCCs) are rare but still present a severe medical problem. These tumors are often disfiguring and impact the quality of life by pain or bleeding. Based on discovery of the hedgehog (Hh) signaling pathway and it’s role in pathogenesis of BCCs, Smoothened (SMO) inhibitors have been developed with Sonidegib being the 2nd in class. It is the only Hh pathway inhibitor investigated in a randomized trial accompanied by pharmacodynamic investigations. Also, the disease assessment criteria applied were more stringent than those used in trials of 1st developed SMO inhibitor – vismodegib, and required annotated photographs, MRI as well as multiple biopsies in case of regression. -

Imatinib (Gleevec™)
Biologicals What Are They? When Did All of this Happen? There are Clear Benefits. Are there also Risks? Brian J Ward Research Institute of the McGill University Health Centre Global Health, Immunity & Infectious Diseases Grand Rounds – March 2016 Biologicals Biological therapy involves the use of living organisms, substances derived from living organisms, or laboratory-produced versions of such substances to treat disease. National Cancer Institute (USA) What Effects Do Steroids Have on Immune Responses? This is your immune system on high dose steroids projects.accessatlanta.com • Suppress innate and adaptive responses • Shut down inflammatory responses in progress • Effects on neutrophils, macrophages & lymphocytes • Few problems because use typically short-term Virtually Every Cell and Pathway in Immune System ‘Target-able’ (Influenza Vaccination) Reed SG et al. Nature Medicine 2013 Nakaya HI et al. Nature Immunology 2011 Landscape - 2013 Antisense (30) Cell therapy (69) Gene Therapy (46) Monoclonal Antibodies (308) Recombinant Proteins (93) Vaccines (250) Other (81) http://www.phrma.org/sites/default/files/pdf/biologicsoverview2013.pdf Therapeutic Category Drugs versus Biologics Patented Ibuprofen (Advil™) Generic Ibuprofen BioSimilars/BioSuperiors ? www.drugbank.ca Patented Etanercept (Enbrel™) BioSimilar Etanercept Etacept™ (India) Biologics in Cancer Therapy Therapeutic Categories • Hormonal Therapy • Monoclonal antibodies • Cytokine therapy • Classical vaccine strategies • Adoptive T-cell or dendritic cells transfer • Oncolytic -

Manufacturer Patient Assistance Programs
Manufacturer Patient Assistance Programs * Provisional Bridging Programs August 2021 highlighted below in blue For medications currently not funded per the BC Cancer Benefit Drug List - http://www.bccancer.bc.ca/systemic-therapy-site/documents/policy%20and%20forms/benefit%20drug%20list.pdf Disclaimer: BC Cancer intends to keep the information on this document as up to date as possible but cannot guarantee that the programs are all available as listed. Contact your local Drug Access Navigator (DAN) for more information UPDATES: New MPAP/Compassionate Access Open: Abiraterone-JAMP, Abiraterone-Sandoz, Everolimus-Sandoz, Gefitinib-JAMP, Gefinitib-Sandoz, Imatinib-JAMP, Encorafenib (Braftovi), Binimetinib (Mektovi), Pralsetinib (Gavreto), Trastuzumab Deruxtecan (Enhertu) Provisional Bridging/Compassionate Access Closed: MPAP Closed: Additional Updates: Drug Manufacturer / PAP Name Contact Information Support Offered Route Strength DIN Abemaciclib (Verzenio) Lilly Phone 1.855.545.5922 Compassionate supply may be available PO 50mg 02487098 * Provisional Bridging Program Lilly Patient Support Program Fax 1.844.503.7749 Financial assistance for patients with or without private insurance may be available PO 100mg 02487101 Email [email protected] PO 150mg 02487128 Web PO 200mg 02487136 Abiraterone (Zytiga) Janssen Phone 1.844.511.2616 Compassionate supply may be available PO 250mg 02371065 * Provisional Bridging Program Janssen BioAdvance Patient Assistance Program Fax 1.855.629.7100 Financial assistance for patients with or without private -

Assessment Report
25 June 2015 EMA/472165/2015 Committee for Medicinal Products for Human Use (CHMP) Assessment report Odomzo International non-proprietary name: sonidegib Procedure No. EMEA/H/C/002839/0000 Note Assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted. 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact An agency of the European Union © European Medicines Agency, 2015. Reproduction is authorised provided the source is acknowledged. Table of contents 1. Background information on the procedure ............................................ 7 1.1. Submission of the dossier ...................................................................................... 7 1.2. Manufacturers ...................................................................................................... 8 1.3. Steps taken for the assessment of the product ......................................................... 8 2. Scientific discussion .............................................................................. 9 2.1. Introduction......................................................................................................... 9 2.2. Quality aspects .................................................................................................. 11 2.2.1. Introduction .................................................................................................... 11 -

Cyclin-Dependent Kinase Inhibitors in Brain Cancer: Current State and Future Directions
Juric et al. Cancer Drug Resist 2020;3:48-62 Cancer DOI: 10.20517/cdr.2019.105 Drug Resistance Review Open Access Cyclin-dependent kinase inhibitors in brain cancer: current state and future directions Viktorija Juric, Brona Murphy Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin D02, Ireland. Correspondence to: Dr. Brona Murphy, Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, 39A York Street, Dublin D02, Ireland. E-mail: [email protected] How to cite this article: Juric V, Murphy B. Cyclin-dependent kinase inhibitors in brain cancer: current state and future directions. Cancer Drug Resist 2020;3:48-62. http://dx.doi.org/10.20517/cdr.2019.105 Received: 5 Nov 2019 First Decision: 4 Dec 2019 Revised: 11 Dec 2019 Accepted: 20 Dec 2019 Published: 19 Mar 2020 Science Editor: Lee M. Graves Copy Editor: Jing-Wen Zhang Production Editor: Jing Yu Abstract Cyclin-dependent kinases (CDKs) are important regulatory enzymes in the normal physiological processes that drive cell-cycle transitions and regulate transcription. Virtually all cancers harbour genomic alterations that lead to the constitutive activation of CDKs, resulting in the proliferation of cancer cells. CDK inhibitors (CKIs) are currently in clinical use for the treatment of breast cancer, combined with endocrine therapy. In this review, we describe the potential of CKIs for the treatment of cancer with specific focus on glioblastoma (GBM), the most common and aggressive primary brain tumour in adults. Despite intense effort to combat GBM with surgery, radiation and temozolomide chemotherapy, the median survival for patients is 15 months and the majority of patients experience disease recurrence within 6-8 months of treatment onset. -

Standard Oncology Criteria C16154-A
Prior Authorization Criteria Standard Oncology Criteria Policy Number: C16154-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DATE DUE BEFORE 03/2016 12/2/2020 1/26/2022 HCPCS CODING TYPE OF CRITERIA LAST P&T APPROVAL/VERSION N/A RxPA Q1 2021 20210127C16154-A PRODUCTS AFFECTED: See dosage forms DRUG CLASS: Antineoplastic ROUTE OF ADMINISTRATION: Variable per drug PLACE OF SERVICE: Retail Pharmacy, Specialty Pharmacy, Buy and Bill- please refer to specialty pharmacy list by drug AVAILABLE DOSAGE FORMS: Abraxane (paclitaxel protein-bound) Cabometyx (cabozantinib) Erwinaze (asparaginase) Actimmune (interferon gamma-1b) Calquence (acalbrutinib) Erwinia (chrysantemi) Adriamycin (doxorubicin) Campath (alemtuzumab) Ethyol (amifostine) Adrucil (fluorouracil) Camptosar (irinotecan) Etopophos (etoposide phosphate) Afinitor (everolimus) Caprelsa (vandetanib) Evomela (melphalan) Alecensa (alectinib) Casodex (bicalutamide) Fareston (toremifene) Alimta (pemetrexed disodium) Cerubidine (danorubicin) Farydak (panbinostat) Aliqopa (copanlisib) Clolar (clofarabine) Faslodex (fulvestrant) Alkeran (melphalan) Cometriq (cabozantinib) Femara (letrozole) Alunbrig (brigatinib) Copiktra (duvelisib) Firmagon (degarelix) Arimidex (anastrozole) Cosmegen (dactinomycin) Floxuridine Aromasin (exemestane) Cotellic (cobimetinib) Fludara (fludarbine) Arranon (nelarabine) Cyramza (ramucirumab) Folotyn (pralatrexate) Arzerra (ofatumumab) Cytosar-U (cytarabine) Fusilev (levoleucovorin) Asparlas (calaspargase pegol-mknl Cytoxan (cyclophosphamide) -

WO 2018/218208 Al 29 November 2018 (29.11.2018) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/218208 Al 29 November 2018 (29.11.2018) W !P O PCT (51) International Patent Classification: Fabio; c/o Bruin Biosciences, Inc., 10225 Barnes Canyon A61K 31/00 (2006.01) A61K 47/00 (2006.01) Road, Suite A104, San Diego, California 92121-2734 (US). BEATON, Graham; c/o Bruin Biosciences, Inc., 10225 (21) International Application Number: Barnes Canyon Road, Suite A104, San Diego, California PCT/US20 18/034744 92121-2734 (US). RAVULA, Satheesh; c/o Bruin Bio (22) International Filing Date: sciences, Inc., 10225 Barnes Canyon Road, Suite A l 04, San 25 May 2018 (25.05.2018) Diego, Kansas 92121-2734 (US). (25) Filing Language: English (74) Agent: MALLON, Joseph J.; 2040 Main Street, Four teenth Floor, Irvine, California 92614 (US). (26) Publication Langi English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/5 11,895 26 May 2017 (26.05.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/5 11,898 26 May 2017 (26.05.2017) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicants: BRUIN BIOSCIENCES, INC. [US/US]; DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, 10225 Barnes Canyon Road, Suite A104, San Diego, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, California 92121-2734 (US). -

Comparator Report on Patient Access to Cancer Medicines in Europe Revisited
COMPARATOR REPORT ON PATIENT ACCESS TO CANCER MEDICINES IN EUROPE REVISITED BENGT JÖNSSON THOMAS HOFMARCHER PETER LINDGREN IHE REPORT NILS WILKING 2016:4 COMPARATOR REPORT ON PATIENT ACCESS TO CANCER MEDICINES IN EUROPE REVISITED Authors: Bengt Jönsson, PhD, professor emeritus Stockholm School of Economics, Stockholm, Sweden Thomas Hofmarcher, MSc The Swedish Institute for Health Economics, Lund Sweden Lund University, Lund, Sweden Peter Lindgren, PhD, associate professor The Swedish Institute for Health Economics, Lund, Sweden Karolinska Institutet, Stockholm, Sweden Nils Wilking, MD, PhD, associate professor Senior strategic advisor cancer, Skåne University Hospital Lund/Malmö, Sweden Karolinska Institutet, Stockholm, Sweden IHE Report 2016:4 e-ISSN 1651-8187 The report can be downloaded from IHE’s website www.ihe.se Please cite this report as: Jönsson, B., Hofmarcher, T., Lindgren, P., Wilking, N. Comparator report on patient access to cancer medicines in Europe revisited. IHE Report 2016:4, IHE: Lund. ACCESS TO CANCER MEDICINES IN EUROPE Content Content 2 Foreword 5 List of Abbreviations 7 Executive summary 9 1 The burden and cost of cancer in Europe 1995–2014 12 1.1 Health burden of cancer 13 1.1.1 Incidence 15 1.1.2 Mortality 19 1.1.3 Survival 25 1.1.4 Burden of disease 27 Economic burden of cancer 31 Direct cost of cancer 35 1.3.1 Comparison with previous results 40 1.3.2 Development of the direct cost over time 41 1.3.3 Spending on cancer and patient outcomes 46 Cost of cancer drugs 48 1.4.1 Development of the cost of cancer drugs