Investigator Initiated Study IRB-29839 an Open-Label Pilot Study To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Potential High-Impact Interventions Report Priority Area 02: Cancer
AHRQ Healthcare Horizon Scanning System – Potential High-Impact Interventions Report Priority Area 02: Cancer Prepared for: Agency for Healthcare Research and Quality U.S. Department of Health and Human Services 540 Gaither Road Rockville, MD 20850 www.ahrq.gov Contract No. HHSA290201000006C Prepared by: ECRI Institute 5200 Butler Pike Plymouth Meeting, PA 19462 December 2012 Statement of Funding and Purpose This report incorporates data collected during implementation of the Agency for Healthcare Research and Quality (AHRQ) Healthcare Horizon Scanning System by ECRI Institute under contract to AHRQ, Rockville, MD (Contract No. HHSA290201000006C). The findings and conclusions in this document are those of the authors, who are responsible for its content, and do not necessarily represent the views of AHRQ. No statement in this report should be construed as an official position of AHRQ or of the U.S. Department of Health and Human Services. This report’s content should not be construed as either endorsements or rejections of specific interventions. As topics are entered into the System, individual topic profiles are developed for technologies and programs that appear to be close to diffusion into practice in the United States. Those reports are sent to various experts with clinical, health systems, health administration, and/or research backgrounds for comment and opinions about potential for impact. The comments and opinions received are then considered and synthesized by ECRI Institute to identify interventions that experts deemed, through the comment process, to have potential for high impact. Please see the methods section for more details about this process. This report is produced twice annually and topics included may change depending on expert comments received on interventions issued for comment during the preceding 6 months. -

Genomic Oncology: Moving Beyond the Tip of the Iceberg Jane De Lartigue, Phd
FeatureCommunity Report Genomic oncology: moving beyond the tip of the iceberg Jane de Lartigue, PhD istorically, cancer has been diagnosed and in patients with lung cancer, even the most efec- treated on the basis of the tissue of ori- tive targeted therapies can fail if used in the wrong Hgin. Te promise of personalized therapy, patient population.5,6 matched more precisely to an individual’s tumor, In recognition of this issue, the oncology feld has was ushered in with the development of molecularly developed molecular biomarkers that can predict targeted therapies, based on a greater understanding response, or lack thereof, to targeted therapy. Drugs of cancer as a genomic-driven disease. Here, we dis- are now commonly being evaluated in trials that cuss some of the evolution of genomic oncology, the select eligible patients on the basis of biomarker pos- inherent complexities and challenges, and how novel itivity, and a number of companion diagnostics have clinical trial designs are among the strategies being been codeveloped to assist in these eforts (Table 1). developed to address them and shape the future of Notable successes include the development of the personalized medicine in cancer. monoclonal antibody trastuzumab for patients with breast cancers that have human epidermal growth The evolution of genomic oncology factor receptor 2 (HER2) gene amplifcation or In the 15 years since the frst map of the human HER2 protein overexpression,7 and the small mol- genome emerged, genetics has become an inte- ecule BRAF kinase inhibitor -
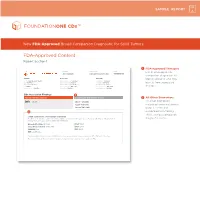
FDA-Approved Content Report Section 1
SAMPLE REPORT New FDA-Approved Broad Companion Diagnostic for Solid Tumors FDA-Approved Content Report Section 1 1 FDA-Approved Therapies PATIENT TUMOR TYPE TRF# List of FDA-approved Jane Sample Lung adenocarcinoma TRFXXXXXX companion diagnostics to PATIENT PHYSICIAN SPECIMEN identify patients who may DISEASE Lung adenocarcinoma ORDERING PHYSICIAN Not Given SPECIMEN SITE Not Given NAME Not Given MEDICAL FACILITY Not Given SPECIMEN ID Not Given benefi t from associated DATE OF BIRTH Not Given ADDITIONAL RECIPIENT Not Given SPECIMEN TYPE Not Given SEX Female MEDICAL FACILITY ID Not Given DATE OF COLLECTION Not Given therapies MEDICAL RECORD # Not Given PATHOLOGIST Not Given SPECIMEN RECEIVED Not Given CDx Associated Findings 1 GENOMIC FINDINGS DETECTED FDA-APPROVED THERAPEUTIC OPTIONS 2 All Other Biomarkers EGFR L858R Gilotrif® (Afatinib) All other biomarkers, Iressa® (Gefitinib) including tumor mutational Tarceva® (Erlotinib) burden (TMB) and 2 microsatellite instability (MSI), without companion OTHER ALTERATIONS & BIOMARKERS IDENTIFIED Results reported in this section are not prescriptive or conclusive for labeled use of any specific therapeutic product. See diagnostic claims professional services section for additional information. Microsatellite Status MS-Stable PTCH1 T416S Tumor Mutation Burden 11 Muts/Mb RBM10 Q494* CDKN2A/B loss TP53 R267P EGFR amplification § Refer to appendix for limitation statements related to detection of any copy number alterations, gene rearrangements, MSI or TMB result in this section. Please refer to appendix -

Overcoming the Challenges of Oral Oncolytic Therapies with a Specialized Crew
Navigating Safely Through Uncharted Waters: Overcoming the Challenges of Oral Oncolytic Therapies with a Specialized Crew Mitchell E. Hughes, PharmD, BCPS, BCOP Clinical Pharmacy Specialist-Hematology/Oncology The Abramson Cancer Center for Advanced Medicine at Penn Medicine Objectives At the completion of this activity, the participant will be able to: 1. List risks associated with dispensing oral oncolytic agent 2. Recognize potential barriers to implementation of a vigilance program for oral oncolytic agents 3. Discuss strategies to improve safety and communications involved with dispensing oral oncolytic agents 2 Disclosure “I have not received any commercial or financial support for this program” 3 Oral Chemotherapy Definition “Any drug you take by mouth to treat cancer. Oral chemo is not given to you with a needle. It’s a liquid or pill that you swallow.” “Chemo you swallow is as strong as other forms of chemo and works just as well. You take oral chemo at home” “But oral chemo drugs cost a lot” –The American Cancer Society 4 Image available from: https://localtvwiti.files.wordpress.com/2014/03/chemo-pills.jpg?quality=85&strip=all https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/chemotherapy/oral-chemotherapy.html Misconceptions Oral chemotherapy is less toxic than intravenous (IV) chemotherapy Oral chemotherapy requires less monitoring than IV Patients will be able to start therapy the day oral chemotherapy is prescribed Oral chemotherapy does not involve any hazardous precautions 5 Image available -

An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib Christina Danial, Kavita Y
Published OnlineFirst November 6, 2015; DOI: 10.1158/1078-0432.CCR-15-1588 Clinical Trial Brief Report Clinical Cancer Research An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib Christina Danial, Kavita Y. Sarin, Anthony E. Oro, and Anne Lynn S. Chang Abstract Purpose: To assess the tumor response to the smoothened sive disease with sonidegib. Three patients experienced stable (SMO) inhibitor, sonidegib (LDE225), in patients with an disease and discontinued sonidegib either due to adverse events advanced basal cell carcinoma (BCC) resistant to treatment with (n ¼ 1) or due to election for surgery (n ¼ 2). The response of one vismodegib (GDC0449). patient was not evaluable. SMO mutations with in vitro data Experimental Design: Nine patients with an advanced suggesting resistance to Hh pathway inhibition were identified BCC that was previously resistant to treatment with vismode- in 5 patients, and none of these patients experienced responses gib were given sonidegib in this investigational, open- while on sonidegib. label study. Tumor response was determined using the Conclusion: Patients with advanced BCCs that were response evaluation criteria in solid tumors. SMO mutations previously resistant to treatment with vismodegib similarly were identified using biopsy samples from the target BCC demonstrated treatment resistance with sonidegib. Patients location. who have developed treatment resistance to an SMO inhibitor Results: The median duration of treatment with sonidegib was may continue to experience tumor progression in response to 6 weeks (range, 3–58 weeks). Five patients experienced progres- other SMO inhibitors. Clin Cancer Res; 1–5. Ó2015 AACR. Introduction Sonidegib (LDE225) is a new SMO inhibitor approved in 2015 by the FDA for locally advanced BCCs. -

Combination of Ponatinib with Hedgehog Antagonist Vismodegib for Therapy-Resistant BCR-ABL1–Positive Leukemia
Published OnlineFirst January 14, 2013; DOI: 10.1158/1078-0432.CCR-12-1777 Clinical Cancer Cancer Therapy: Preclinical Research See related commentary by Dao and Tyner, p. 1309 Combination of Ponatinib with Hedgehog Antagonist Vismodegib for Therapy-Resistant BCR-ABL1–Positive Leukemia Seiichiro Katagiri1, Tetsuzo Tauchi1, Seiichi Okabe1, Yosuke Minami2, Shinya Kimura3, Taira Maekawa4, Tomoki Naoe2, and Kazuma Ohyashiki1 Abstract Purpose: The Hedgehog signaling pathway is a key regulator of cell growth and differentiation during development. Whereas the Hedgehog pathway is inactive in most normal adult tissues, Hedgehog pathway reactivation has been implicated in the pathogenesis of several neoplasms including BCR-ABL1–positive leukemia. The clear link between the Hedgehog pathway and BCR-ABL1–positive leukemia led to an effort to identify small molecules to block the pathway. Experimental Design: We investigated the combined effects of vismodegib and ponatinib, a pan-ABL1 kinase inhibitor, in nonobese diabetic/severe-combined immunodeficiency (NOD/SCID) repopulating T315I BCR-ABL1–positive cells in vitro and in vivo. Results: We observed that combination with vismodegib and ponatinib helps to eliminate therapy- resistant NOD/SCID repopulating T315I BCR-ABL1–positive cells. The percentage of CD19-positive leukemia cells in peripheral blood was significantly lower in vismodegib þ ponatinib–treated mice than that of the vehicle or ponatinib alone (P < 0.001). Spleen weights were also lower in vismodegib þ ponatinib–treated mice than in ponatinib alone (P < 0.05). Overall tumor burden, as assessed by BCR-ABL mRNA from bone marrow cells, was significantly lower in vismodegib þ ponatinib–treated mice than in ponatinib alone (P < 0.005). -

Imatinib (Gleevec™)
Biologicals What Are They? When Did All of this Happen? There are Clear Benefits. Are there also Risks? Brian J Ward Research Institute of the McGill University Health Centre Global Health, Immunity & Infectious Diseases Grand Rounds – March 2016 Biologicals Biological therapy involves the use of living organisms, substances derived from living organisms, or laboratory-produced versions of such substances to treat disease. National Cancer Institute (USA) What Effects Do Steroids Have on Immune Responses? This is your immune system on high dose steroids projects.accessatlanta.com • Suppress innate and adaptive responses • Shut down inflammatory responses in progress • Effects on neutrophils, macrophages & lymphocytes • Few problems because use typically short-term Virtually Every Cell and Pathway in Immune System ‘Target-able’ (Influenza Vaccination) Reed SG et al. Nature Medicine 2013 Nakaya HI et al. Nature Immunology 2011 Landscape - 2013 Antisense (30) Cell therapy (69) Gene Therapy (46) Monoclonal Antibodies (308) Recombinant Proteins (93) Vaccines (250) Other (81) http://www.phrma.org/sites/default/files/pdf/biologicsoverview2013.pdf Therapeutic Category Drugs versus Biologics Patented Ibuprofen (Advil™) Generic Ibuprofen BioSimilars/BioSuperiors ? www.drugbank.ca Patented Etanercept (Enbrel™) BioSimilar Etanercept Etacept™ (India) Biologics in Cancer Therapy Therapeutic Categories • Hormonal Therapy • Monoclonal antibodies • Cytokine therapy • Classical vaccine strategies • Adoptive T-cell or dendritic cells transfer • Oncolytic -

Access to Cancer Medicines in Australia
Access to cancer medicines in Australia Medicines Australia Oncology Industry Taskforce July 2013 Contents Glossary ..................................................................................................................................... i Executive summary .................................................................................................................... i 1 Background ..................................................................................................................... 1 1.1 Purpose of this report ....................................................................................................... 2 1.2 Methods ........................................................................................................................... 3 1.3 Report structure ............................................................................................................... 9 2 Cancer in Australia and other countries ......................................................................... 10 2.1 Population statistics on cancer ........................................................................................ 10 2.2 Population impacts of cancer in Australia ........................................................................ 24 2.3 Summary ........................................................................................................................ 33 3 Current and future cancer medicines ............................................................................ 34 3.1 Current -

Session #3 Mfoflox-6 FOLFIRI ± Bevacizumab
Management of Patients Receiving Oral Chemotherapy Siu Fun Wong, PharmD, FASHP, FCSHP Professor and Associate Dean Chapman University School of Pharmacy (CUSP) Irvine, CA Disclosure I have no conflict of interest . Objectives At the end of the presentation, the participants will be able to: 1. Recognize the difference in care required by patients receiving parenteral vs. oral chemotherapy 2. Deliver personalized care to patients receiving oral chemotherapy 3. Provide appropriate education relating to oral chemotherapy to patients and/or caregivers. Types of Oral Anti-cancer Chemotherapy Cytotoxic Agents Hormone Agents Immunomodulators Differentiating Agents Targeted Therapy Paradigm Change The rise in the use of oral oncology drugs represents a major shift in the management of patient . Directly observed treatment to unsupervised self- administration setting . Intermittent to daily chronic therapy . Perception of less toxicity with oral route of administration – both patients and providers1 . Patient preference due to convenience without compromise for efficacy2 . Provider preference, especially in palliative or adjuvant setting where QOL is paramount3 1Sharma S & Saltz LB. The Oncologist 2000; 5:99-107 2 Liu G et al. J Clin Oncol1997; 15(1):110-115 3 Benjamin et al. Eur J Cancer 2012;48(6):912-920 Goal How to empower patients to take the right dose at the right time under the right circumstances and to complete the treatment !! Effective Use and Monitoring Key (Unique) Elements to Consider Drug Administration Drug Interactions – Pharmacogenomics -

Making Models More Accessible to Decision Makers
11th November 2014 Making models more accessible to decision makers Michael Barry, PhD National Centre for Pharmacoeconomics, Dublin 11th November 2014 The NCPE conducts the health technology assessment (HTA) of pharmaceutical products for the Health Service Executive Recommendations on 192 medicines for 208 indications 3rd November 2014 1 11th November 2014 The HTA process The process begins with the price Rapid review to application by the determine whether a manufacturer full HTA is required Full HTA with a 90 day time frame NCPE submission to the HSE – CPU. This report will be considered by the New Drugs Group Committee 11th November 2014 Economic evaluations conducted in the Irish healthcare setting are usually in the form of CEA or CUA. Cost-effectiveness analysis (CEA) e.g. COST/LYG Cost-utility analysis (CUA) e.g. COST/QALY 2 11th November 2014 Cost-effectiveness threshold The line passing through the origin represents our ‘acceptable’ cost- effectiveness ratio. That is our maximum (or threshold) willingness-to-pay for a unit of effect ( life year or QALY). Cost (€) Q4 Q1 Effect (QALY) Q3 Q2 The QALY threshold to be used in the HTA process is € 45,000/QALY 11th November 2014 Number of products reviewed by the NCPE 2006 - 2014 3 11th November 2014 We will read through the HTA submission ……carefully ! 11th November 2014 Products reviewed in 2014 • HTA not required = 24 (44%) • Reimbursement recommended = 5 (9%) • Full HTA required = 12 (22%) • Reimbursement not recommended = 13 (24%) 5 at the submitted price & 7 oncology products -

Manufacturer Patient Assistance Programs
Manufacturer Patient Assistance Programs * Provisional Bridging Programs August 2021 highlighted below in blue For medications currently not funded per the BC Cancer Benefit Drug List - http://www.bccancer.bc.ca/systemic-therapy-site/documents/policy%20and%20forms/benefit%20drug%20list.pdf Disclaimer: BC Cancer intends to keep the information on this document as up to date as possible but cannot guarantee that the programs are all available as listed. Contact your local Drug Access Navigator (DAN) for more information UPDATES: New MPAP/Compassionate Access Open: Abiraterone-JAMP, Abiraterone-Sandoz, Everolimus-Sandoz, Gefitinib-JAMP, Gefinitib-Sandoz, Imatinib-JAMP, Encorafenib (Braftovi), Binimetinib (Mektovi), Pralsetinib (Gavreto), Trastuzumab Deruxtecan (Enhertu) Provisional Bridging/Compassionate Access Closed: MPAP Closed: Additional Updates: Drug Manufacturer / PAP Name Contact Information Support Offered Route Strength DIN Abemaciclib (Verzenio) Lilly Phone 1.855.545.5922 Compassionate supply may be available PO 50mg 02487098 * Provisional Bridging Program Lilly Patient Support Program Fax 1.844.503.7749 Financial assistance for patients with or without private insurance may be available PO 100mg 02487101 Email [email protected] PO 150mg 02487128 Web PO 200mg 02487136 Abiraterone (Zytiga) Janssen Phone 1.844.511.2616 Compassionate supply may be available PO 250mg 02371065 * Provisional Bridging Program Janssen BioAdvance Patient Assistance Program Fax 1.855.629.7100 Financial assistance for patients with or without private -

Cell Plasticity and Prostate Cancer: the Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance
cancers Review Cell Plasticity and Prostate Cancer: The Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance Sofia Papanikolaou, Aikaterini Vourda, Spyros Syggelos and Kostis Gyftopoulos * Department of Anatomy, University of Patras School of Medicine, Rion, 26504 Patras, Greece; [email protected] (S.P.); [email protected] (A.V.); [email protected] (S.S.) * Correspondence: [email protected] Simple Summary: Although epithelial-to-mesenchymal transition (EMT) is a well-known cellular process involved during normal embryogenesis and wound healing, it also has a dark side; it is a complex process that provides tumor cells with a more aggressive phenotype, facilitating tumor metastasis and even resistance to therapy. This review focuses on the key pathways of EMT in the pathogenesis of prostate cancer and the development of metastases and evasion of currently available treatments. Abstract: Prostate cancer, the second most common malignancy in men, is characterized by high heterogeneity that poses several therapeutic challenges. Epithelial–mesenchymal transition (EMT) is a dynamic, reversible cellular process which is essential in normal embryonic morphogenesis and Citation: Papanikolaou, S.; Vourda, wound healing. However, the cellular changes that are induced by EMT suggest that it may also A.; Syggelos, S.; Gyftopoulos, K. Cell play a central role in tumor progression, invasion, metastasis, and resistance to current therapeutic Plasticity and Prostate Cancer: The options. These changes include enhanced motility and loss of cell–cell adhesion that form a more Role of Epithelial–Mesenchymal aggressive cellular phenotype. Moreover, the reverse process (MET) is a necessary element of the Transition in Tumor Progression, Invasion, Metastasis and Cancer metastatic tumor process.