Cyclin-Dependent Kinase Inhibitors in Brain Cancer: Current State and Future Directions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Investigator Initiated Study IRB-29839 an Open-Label Pilot Study To
Investigator Initiated Study IRB-29839 An open-label pilot study to evaluate the efficacy and safety of a combination treatment of Sonidegib and BKM120 for the treatment of advanced basal cell carcinomas Version 05 September 2016 NCT02303041 DATE: 12Dec2018 1 Coordinating Center Stanford Cancer Center 875 Blake Wilbur Drive Stanford, CA 94305 And 450 Broadway, MC 5334 Redwood City, CA 94603 Protocol Director and Principal Investigator Anne Lynn S Chang, MD, Director of Dermatological Clinical Trials 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] Co-Investigator Anthony Oro, MD PhD 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] Biostatistician Shufeng Li, MS 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] Study Coordinator Ann Moffat 450 Broadway St, MC 5334 Redwood City, CA 94603 [email protected] 2 Table of Contents 1 Background ................................................................. 7 1.1 Disease Background ..................................................... 7 1.2 Hedgehog Pathway and mechanism of action ............................... 7 1.3 PI3K Pathway and mechanism of action ................................... 9 1.4 Sonidegib Compound Information ............ Error! Bookmark not defined. 1.4.1 Preclinical Studies for Sonidegib ....................................................................11 1.4.2 Muscular system...............................................................................................13 1.4.3 Skeletal system ................................................................................................13 -

Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin-Dependent Kinase 2 and AKT Inhibition
Published OnlineFirst September 23, 2016; DOI: 10.1158/1078-0432.CCR-16-0620 Biology of Human Tumors Clinical Cancer Research Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin- Dependent Kinase 2 and AKT Inhibition George Au-Yeung1,2, Franziska Lang1, Walid J. Azar1, Chris Mitchell1, Kate E. Jarman3, Kurt Lackovic3,4, Diar Aziz5, Carleen Cullinane1,6, Richard B. Pearson1,2,7, Linda Mileshkin2,8, Danny Rischin2,8, Alison M. Karst9, Ronny Drapkin10, Dariush Etemadmoghadam1,2,5, and David D.L. Bowtell1,2,7,11 Abstract Purpose: Cyclin E1 (CCNE1) amplification is associated with Results: We validate CDK2 as a therapeutic target by demon- primary treatment resistance and poor outcome in high-grade strating selective sensitivity to gene suppression. However, we found serous ovarian cancer (HGSC). Here, we explore approaches to that dinaciclib did not trigger amplicon-dependent sensitivity in a target CCNE1-amplified cancers and potential strategies to over- panel of HGSC cell lines. A high-throughput compound screen come resistance to targeted agents. identified synergistic combinations in CCNE1-amplified HGSC, Experimental Design: To examine dependency on CDK2 in including dinaciclib and AKT inhibitors. Analysis of genomic data CCNE1-amplified HGSC, we utilized siRNA and conditional from TCGA demonstrated coamplification of CCNE1 and AKT2. shRNA gene suppression, and chemical inhibition using dina- Overexpression of Cyclin E1 and AKT isoforms, in addition to ciclib, a small-molecule CDK2 inhibitor. High-throughput mutant TP53, imparted malignant characteristics in untransformed compound screening was used to identify selective synergistic fallopian tube secretory cells, the dominant site of origin of HGSC. -

A Study of Abemaciclib (LY2835219)
A Study of Abemaciclib (LY2835219) in Combination With Temozolomide and Irinotecan and Abemaciclib in Combination With Temozolomide in Children and Young Adult Participants With Solid Tumors Status: Not yet recruiting Eligibility Criteria Sex: All Age: up to 18 Years old This study is NOT accepting healthy volunteers Inclusion Criteria: • Body weight ≥10 kilograms and body surface area (BSA) ≥0.5 meters squared. • Participants with any relapsed/refractory malignant solid tumor (excluding lymphoma), including central nervous system tumors, that have progressed on standard therapies and, in the judgment of the investigator, are appropriate candidates for the experimental therapy combination in the study part that is currently enrolling. • Participants must have at least one measurable (per Response Criteria in Solid Tumors [RECIST v1.1; [Eisenhauer et al. 2009] or Response Assessment in Neuro-Oncology (RANO) for central nervous system (CNS) tumors [Wen et al. 2010]) or evaluable lesion. • Participants must have had histologic verification of malignancy at original diagnosis or relapse, except: • Participants with extra-cranial germ-cell tumors who have elevations of serum tumor markers including alpha-fetoprotein or beta- human chorionic gonadotropin (HCG). • Participants with intrinsic brain stem tumors or participants with CNS-germ cell tumors and elevations of CSF or serum tumor markers including alpha-fetoprotein or beta-HCG. • A Lansky score ≥50 for participants ≤16 years of age or Karnofsky score ≥50 for participants >16 years of age. • Participants must have discontinued all previous treatments for cancer or investigational agents and must have recovered from the acute effects to Grade ≤1 at the time of enrollment. -

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors
Published OnlineFirst May 23, 2016; DOI: 10.1158/2159-8290.CD-16-0095 RESEARCH ARTICLE Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors Amita Patnaik1, Lee S. Rosen2, Sara M. Tolaney3, Anthony W. Tolcher1, Jonathan W. Goldman2, Leena Gandhi3, Kyriakos P. Papadopoulos1, Muralidhar Beeram1, Drew W. Rasco1, John F. Hilton3, Aejaz Nasir4, Richard P. Beckmann4, Andrew E. Schade4, Angie D. Fulford4, Tuan S. Nguyen4, Ricardo Martinez4, Palaniappan Kulanthaivel4, Lily Q. Li4, Martin Frenzel4, Damien M. Cronier4, Edward M. Chan4, Keith T. Flaherty5, Patrick Y. Wen3, and Geoffrey I. Shapiro3 ABSTRACT We evaluated the safety, pharmacokinetic profile, pharmacodynamic effects, and antitumor activity of abemaciclib, an orally bioavailable inhibitor of cyclin-dependent kinases (CDK) 4 and 6, in a multicenter study including phase I dose escalation followed by tumor- specific cohorts for breast cancer, non–small cell lung cancer (NSCLC), glioblastoma, melanoma, and colorectal cancer. A total of 225 patients were enrolled: 33 in dose escalation and 192 in tumor-specific cohorts. Dose-limiting toxicity was grade 3 fatigue. The maximum tolerated dose was 200 mg every 12 hours. The most common possibly related treatment-emergent adverse events involved fatigue and the gastrointestinal, renal, or hematopoietic systems. Plasma concentrations increased with dose, and pharmacodynamic effects were observed in proliferating keratinocytes and tumors. Radiographic responses were achieved in previously treated patients with breast cancer, NSCLC, and melanoma. For hormone receptor–positive breast cancer, the overall response rate was 31%; moreover, 61% of patients achieved either response or stable disease lasting ≥6 months. -

The CDK4/6 Inhibitor LY2835219 Has Potent Activity in Combination with Mtor Inhibitor in Head and Neck Squamous Cell Carcinoma
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 12 The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma Bo Mi Ku1,*, Seong Yoon Yi3,*, Jiae Koh1, Yeon-Hee Bae1, Jong-Mu Sun2, Se-hoon Lee2, Jin Seok Ahn2, Keunchil Park2, Myung-Ju Ahn2 1 Samsung Biomedical Research Institute, Seoul, Korea 2 Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea 3 Division of Hematology-Oncology, Department of Internal Medicine, Inje University Ilsan Paik Hospital, Gyeonggi-do, Korea *These authors contributed equally to this work Correspondence to: Myung-Ju Ahn, e-mail: [email protected] Keywords: head and neck cancer, CDK4/6 inhibitor, mTOR, cell cycle, targeted therapy Received: September 15, 2015 Accepted: January 23, 2016 Published: February 21, 2016 ABSTRACT Deletion of CDKN2A (p16) or amplification ofCCND1 (cyclin D1) occurs commonly in head and neck squamous cell carcinoma (HNSCC) and induces sustained cyclin- dependent kinase (CDK) 4/6 activation. Here, we report the antiproliferative activity of LY2835219, a selective CDK4/6 inhibitor through inhibition of CDK4/6-dependent Ser780 phosphorylation in retinoblastoma (RB) and induction of cell cycle arrest in HNSCC cells. In addition, we demonstrated the antitumor effects of HNSCC xenografts to LY2835219 in vivo. Given the limited effect in HNSCC as a single-agent treatment with LY2835219, a combinational strategy is required to enhance antitumor activity. At the molecular level, we found that LY2835219 inhibited activation of AKT and ERK, but not mTOR. The combination of LY2835219 with mTOR inhibitor was found to be more effective than either drug alone in vitro and in vivo. -

Identification of Candidate Repurposable Drugs to Combat COVID-19 Using a Signature-Based Approach
www.nature.com/scientificreports OPEN Identifcation of candidate repurposable drugs to combat COVID‑19 using a signature‑based approach Sinead M. O’Donovan1,10, Ali Imami1,10, Hunter Eby1, Nicholas D. Henkel1, Justin Fortune Creeden1, Sophie Asah1, Xiaolu Zhang1, Xiaojun Wu1, Rawan Alnafsah1, R. Travis Taylor2, James Reigle3,4, Alexander Thorman6, Behrouz Shamsaei4, Jarek Meller4,5,6,7,8 & Robert E. McCullumsmith1,9* The COVID‑19 pandemic caused by the novel SARS‑CoV‑2 is more contagious than other coronaviruses and has higher rates of mortality than infuenza. Identifcation of efective therapeutics is a crucial tool to treat those infected with SARS‑CoV‑2 and limit the spread of this novel disease globally. We deployed a bioinformatics workfow to identify candidate drugs for the treatment of COVID‑19. Using an “omics” repository, the Library of Integrated Network‑Based Cellular Signatures (LINCS), we simultaneously probed transcriptomic signatures of putative COVID‑19 drugs and publicly available SARS‑CoV‑2 infected cell lines to identify novel therapeutics. We identifed a shortlist of 20 candidate drugs: 8 are already under trial for the treatment of COVID‑19, the remaining 12 have antiviral properties and 6 have antiviral efcacy against coronaviruses specifcally, in vitro. All candidate drugs are either FDA approved or are under investigation. Our candidate drug fndings are discordant with (i.e., reverse) SARS‑CoV‑2 transcriptome signatures generated in vitro, and a subset are also identifed in transcriptome signatures generated from COVID‑19 patient samples, like the MEK inhibitor selumetinib. Overall, our fndings provide additional support for drugs that are already being explored as therapeutic agents for the treatment of COVID‑19 and identify promising novel targets that are worthy of further investigation. -

A Novel CDK9 Inhibitor Increases the Efficacy of Venetoclax (ABT-199) in Multiple Models of Hematologic Malignancies
Leukemia (2020) 34:1646–1657 https://doi.org/10.1038/s41375-019-0652-0 ARTICLE Molecular targets for therapy A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies 1 1 2,3 4 1 5 6 Darren C. Phillips ● Sha Jin ● Gareth P. Gregory ● Qi Zhang ● John Xue ● Xiaoxian Zhao ● Jun Chen ● 1 1 1 1 1 1 Yunsong Tong ● Haichao Zhang ● Morey Smith ● Stephen K. Tahir ● Rick F. Clark ● Thomas D. Penning ● 2,7 3 5 1 4 2,7 Jennifer R. Devlin ● Jake Shortt ● Eric D. Hsi ● Daniel H. Albert ● Marina Konopleva ● Ricky W. Johnstone ● 8 1 Joel D. Leverson ● Andrew J. Souers Received: 27 November 2018 / Revised: 18 October 2019 / Accepted: 13 November 2019 / Published online: 11 December 2019 © The Author(s) 2019 Abstract MCL-1 is one of the most frequently amplified genes in cancer, facilitating tumor initiation and maintenance and enabling resistance to anti-tumorigenic agents including the BCL-2 selective inhibitor venetoclax. The expression of MCL-1 is maintained via P-TEFb-mediated transcription, where the kinase CDK9 is a critical component. Consequently, we developed a series of potent small-molecule inhibitors of CDK9, exemplified by the orally active A-1592668, with CDK selectivity profiles 1234567890();,: 1234567890();,: that are distinct from related molecules that have been extensively studied clinically. Short-term treatment with A-1592668 rapidly downregulates RNA pol-II (Ser 2) phosphorylation resulting in the loss of MCL-1 protein and apoptosis in MCL-1- dependent hematologic tumor cell lines. This cell death could be attenuated by either inhibiting caspases or overexpressing BCL-2 protein. -
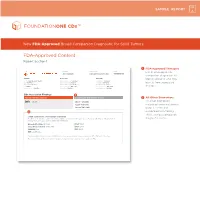
FDA-Approved Content Report Section 1
SAMPLE REPORT New FDA-Approved Broad Companion Diagnostic for Solid Tumors FDA-Approved Content Report Section 1 1 FDA-Approved Therapies PATIENT TUMOR TYPE TRF# List of FDA-approved Jane Sample Lung adenocarcinoma TRFXXXXXX companion diagnostics to PATIENT PHYSICIAN SPECIMEN identify patients who may DISEASE Lung adenocarcinoma ORDERING PHYSICIAN Not Given SPECIMEN SITE Not Given NAME Not Given MEDICAL FACILITY Not Given SPECIMEN ID Not Given benefi t from associated DATE OF BIRTH Not Given ADDITIONAL RECIPIENT Not Given SPECIMEN TYPE Not Given SEX Female MEDICAL FACILITY ID Not Given DATE OF COLLECTION Not Given therapies MEDICAL RECORD # Not Given PATHOLOGIST Not Given SPECIMEN RECEIVED Not Given CDx Associated Findings 1 GENOMIC FINDINGS DETECTED FDA-APPROVED THERAPEUTIC OPTIONS 2 All Other Biomarkers EGFR L858R Gilotrif® (Afatinib) All other biomarkers, Iressa® (Gefitinib) including tumor mutational Tarceva® (Erlotinib) burden (TMB) and 2 microsatellite instability (MSI), without companion OTHER ALTERATIONS & BIOMARKERS IDENTIFIED Results reported in this section are not prescriptive or conclusive for labeled use of any specific therapeutic product. See diagnostic claims professional services section for additional information. Microsatellite Status MS-Stable PTCH1 T416S Tumor Mutation Burden 11 Muts/Mb RBM10 Q494* CDKN2A/B loss TP53 R267P EGFR amplification § Refer to appendix for limitation statements related to detection of any copy number alterations, gene rearrangements, MSI or TMB result in this section. Please refer to appendix -

Cyclacel's CYC065 CDK Inhibitor Demonstrates Synergy With
Cyclacel’s CYC065 CDK Inhibitor Demonstrates Synergy With Venetoclax By Dual Targeting Of Chronic Lymphocytic Leukemia April 17, 2018 Suppression of both BCl-2 and Mcl-1 anti-apoptotic proteins is a novel strategy in CLL BERKELEY HEIGHTS, N.J., April 17, 2018 (GLOBE NEWSWIRE) -- Cyclacel Pharmaceuticals, Inc. (NASDAQ:CYCC) (NASDAQ:CYCCP) ("Cyclacel" or the "Company"), a biopharmaceutical company developing oral therapies that target various phases of cell cycle control for the treatment of cancer and other serious disorders, today announced the presentation by investigators led by William Plunkett, PhD, Professor and Deputy Chair, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, Texas, of preclinical data demonstrating strong synergy between Cyclacel’s CDK2/9 inhibitor, CYC065, and the Bcl-2 inhibitor, venetoclax (ABT-199, AbbVie) in chronic lymphocytic leukemia (CLL) samples obtained from patients. The data were presented at the American Association for Cancer Research (AACR) Annual Meeting being held April 14-18, 2018 in Chicago, Illinois. “The MD Anderson data show that the combination of CYC065 and venetoclax is strongly synergistic in primary CLL cells from patients, including those with 17p deletions. In addition, the combination was active in two CLL samples which were resistant to either agent alone. These findings support the hypothesis that dual targeting of the Mcl-1- and Bcl-2-dependent mechanisms could induce synergistic cell death by apoptosis,” said Spiro Rombotis, President and Chief Executive Officer of Cyclacel. “Last weekend during the same AACR conference, we reported that CYC065 durably suppresses Mcl-1, a member of the Bcl-2 family of survival proteins, in patients with advanced solid tumors1. -

Pharmacologic Properties of AG-012986, a Pan-Cyclin- Dependent Kinase Inhibitor with Antitumor Efficacy
818 Pharmacologic properties of AG-012986, a pan-cyclin- dependent kinase inhibitor with antitumor efficacy Cathy Zhang,1 Karen Lundgren,1 Zhengming Yan,1 optimization of AG-012986 provided guidance for select- Maria E. Arango,1 Sharon Price,1 Andrea Huber,1 ing a treatment schedule to achieve the best antitumor Joseph Higgins,1 Gabriel Troche,1 efficacy while minimizing the risk of adverse side effects. Judith Skaptason,2 Tatiana Koudriakova,2 [Mol Cancer Ther 2008;7(4):818–28] Jim Nonomiya,4 Michelle Yang,5 1 1 1 Patrick O’Connor, Steve Bender, Gerrit Los, Introduction Cristina Lewis,4 and Bart Jessen3 Cyclin-dependent kinases (CDK) and their regulatory Departments of 1Cancer Biology, 2Pharmacokinetics, Dynamics cyclin partners play critical roles in cell cycle control and and Metabolism, 3Drug Safety Research and Development, the regulation of cell transcription. Progression through 4 5 Biochemical Pharmacology, and Medicinal Chemistry, the different stages of cell cycle is governed by the Pfizer Global Research and Development, La Jolla, California activities of the CDK1, CDK2, CDK4, CDK6, and possibly CDK3. CDK4/cyclin D, CDK6/cyclin D, and CDK2/cyclin E Abstract phosphorylate the retinoblastoma (Rb) protein at multiple AG-012986 is a multitargeted cyclin-dependent kinase sites, which results in activation of the E2F family of (CDK) inhibitor active against CDK1, CDK2, CDK4/6, transcription factors and serves as a trigger for cells to CDK5, and CDK9, with selectivity over a diverse panel of advance beyond the G1 checkpoint into S phase (1–3). non-CDK kinases. Here, we report the potent antitumor During S phase, CDK2/cyclin A phosphorylates several efficacies of AG-012986 against multiple tumor lines proteins, including E2F, to regulate progression through in vitro and in vivo. -

Anticancer and Radiosensitizing Effects of the Cyclin-Dependent Kinase Inhibitors, AT7519 and SNS‑032, on Cervical Cancer
INTERNATIONAL JOURNAL OF ONCOLOGY 53: 703-712, 2018 Anticancer and radiosensitizing effects of the cyclin-dependent kinase inhibitors, AT7519 and SNS-032, on cervical cancer MI AE KANG1*, WONWOO KIM2*, HYE-RAM JO1,3, YOUNG-JOO SHIN4, MOON-HONG KIM5 and JAE-HOON JEONG1,3 1Division of Applied Radiation Bioscience, and 2Radiation Non-Clinic Center, Korea Institute of Radiological and Medical Science, Seoul 01812; 3Radiological and Medico-Oncological Sciences, Korea University of Science and Technology, Daejeon 34113; 4Department of Radiation Oncology, Inje University Sanggye Paik Hospital, Seoul 01757; 5Department of Obstetrics and Gynecology, Korea Institute of Radiological and Medical Sciences, Seoul 01812, Republic of Korea Received January 30, 2018; Accepted May 17, 2018 DOI: 10.3892/ijo.2018.4424 Abstract. Cyclin-dependent kinases (CDK) are considered the utilization of AT7519 and SNS-032 as part of an adjuvant to be potential targets of anticancer drugs that can interrupt treatment may help control cervical cancer progression. the uncontrolled division of cancer cells. In this study, we selected two selective CDK inhibitors, AT7519 and SNS-032, Introduction from current clinical trials and examined their anticancer and radiosensitizing effects in a cervical cancer model. SNS-032 Cyclin-dependent kinases (CDKs) are present in all known was found to be more potent than AT7519, with a lower half eukaryotes, and their regulatory functions during the cell cycle maximal inhibitory concentration (IC50) value. Both AT7519 are evolutionarily conserved. Cyclin-CDK complexes phos- and SNS-032 induced the apoptosis, premature senescence and phorylate specific substrates, according to the requirements cytostasis of cervical cancer cells, which led to the attenuation of a particular cell cycle phase.