AKR1C1 Controls Cisplatin-Resistance in Head and Neck Squamous Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene-Expression Signature Regulated by the KEAP1-NRF2-CUL3 Axis Is Associated with a Poor Prognosis in Head and Neck Squamous Cell Cancer Akhileshwar Namani1†, Md
Namani et al. BMC Cancer (2018) 18:46 DOI 10.1186/s12885-017-3907-z RESEARCH ARTICLE Open Access Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer Akhileshwar Namani1†, Md. Matiur Rahaman2†, Ming Chen2* and Xiuwen Tang1* Abstract Background: NRF2 is the key regulator of oxidative stress in normal cells and aberrant expression of the NRF2 pathway due to genetic alterations in the KEAP1 (Kelch-like ECH-associated protein 1)-NRF2 (nuclear factor erythroid 2 like 2)-CUL3 (cullin 3) axis leads to tumorigenesis and drug resistance in many cancers including head and neck squamous cell cancer (HNSCC). The main goal of this study was to identify specific genes regulated by the KEAP1-NRF2-CUL3 axis in HNSCC patients, to assess the prognostic value of this gene signature in different cohorts, and to reveal potential biomarkers. Methods: RNA-Seq V2 level 3 data from 279 tumor samples along with 37 adjacent normal samples from patients enrolled in the The Cancer Genome Atlas (TCGA)-HNSCC study were used to identify upregulated genes using two methods (altered KEAP1-NRF2-CUL3 versus normal, and altered KEAP1-NRF2-CUL3 versus wild-type). We then used a new approach to identify the combined gene signature by integrating both datasets and subsequently tested this signature in 4 independent HNSCC datasets to assess its prognostic value. In addition, functional annotation using the DAVID v6.8 database and protein-protein interaction (PPI) analysis using the STRING v10 databasewereperformedonthesignature. Results: A signature composed of a subset of 17 genes regulated by the KEAP1-NRF2-CUL3 axis was identified by overlapping both the upregulated genes of altered versus normal (251 genes) and altered versus wild-type (25 genes) datasets. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Functional Role of Pentose Phosphate Pathway and Glutamine in Cancer Cell
Functional Role of Pentose Phosphate Pathway and Glutamine in Cancer Cell Ibrahim Halil Polat ADVERTIMENT. La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del servei TDX (www.tdx.cat) i a través del Dipòsit Digital de la UB (diposit.ub.edu) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei TDX ni al Dipòsit Digital de la UB. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a TDX o al Dipòsit Digital de la UB (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA. La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del servicio TDR (www.tdx.cat) y a través del Repositorio Digital de la UB (diposit.ub.edu) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio TDR o al Repositorio Digital de la UB. -

Development of Potent and Selective Inhibitors of Aldo−Keto Reductase
Article pubs.acs.org/jmc Development of Potent and Selective Inhibitors of Aldo−Keto Reductase 1C3 (Type 5 17β-Hydroxysteroid Dehydrogenase) Based on N-Phenyl-Aminobenzoates and Their Structure−Activity Relationships † § ‡ § † † † ‡ Adegoke O. Adeniji, , Barry M. Twenter, , Michael C. Byrns, Yi Jin, Mo Chen, Jeffrey D. Winkler,*, † and Trevor M. Penning*, † Department of Pharmacology and Center of Excellence in Environmental Toxicology, Perelman School of Medicine, University of Pennsylvania, 130C John Morgan Building, 3620 Hamilton Walk, Philadelphia, Pennsylvania 19104-6084, United States ‡ Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104, United States *S Supporting Information ABSTRACT: Aldo−keto reductase 1C3 (AKR1C3; type 5 17β-hydroxy- steroid dehydrogenase) is overexpressed in castration resistant prostate cancer (CRPC) and is implicated in the intratumoral biosynthesis of testosterone and 5α-dihydrotestosterone. Selective AKR1C3 inhibitors are required because compounds should not inhibit the highly related AKR1C1 and AKR1C2 isoforms which are involved in the inactivation of 5α-dihydrotestosterone. NSAIDs, N-phenylanthranilates in particular, are potent but nonselective AKR1C3 inhibitors. Using flufenamic acid, 2-{[3-(trifluoromethyl)phenyl]amino}- benzoic acid, as lead compound, five classes of structural analogues were synthesized and evaluated for AKR1C3 inhibitory potency and selectivity. Structure−activity relationship (SAR) studies revealed that a meta-carboxylic acid group relative to the amine conferred pronounced AKR1C3 selectivity without loss of potency, while electron withdrawing groups on the phenylamino B-ring were optimal for AKR1C3 inhibition. Lead compounds did not inhibit COX-1 or COX-2 but blocked the AKR1C3 mediated production of testosterone in LNCaP-AKR1C3 cells. These compounds offer promising leads toward new therapeutics for CRPC. -

Bioinformatic and Modelling Approaches for a System-Level Understanding of Oxidative Stress Toxicity Elias Zgheib
Bioinformatic and modelling approaches for a system-level understanding of oxidative stress toxicity Elias Zgheib To cite this version: Elias Zgheib. Bioinformatic and modelling approaches for a system-level understanding of oxidative stress toxicity. Quantitative Methods [q-bio.QM]. Université de Technologie de Compiègne, 2018. English. NNT : 2018COMP2464. tel-02088169 HAL Id: tel-02088169 https://tel.archives-ouvertes.fr/tel-02088169 Submitted on 2 Apr 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Par Elias ZGHEIB Bioinformatic and modelling approaches for a system- level understanding of oxidative stress toxicity Thèse présentée pour l’obtention du grade de Docteur de l’UTC Soutenue le 18 décembre 2018 Spécialité : Bio-ingénierie et Mathématiques Appliquées : Unité de Recherche Biomécanique et Bio-ingénierie (UMR-7338) D2464 BIOINFORMATIC AND MODELLING APPROACHES FOR A SYSTEM-LEVEL UNDERSTANDING OF OXIDATIVE STRESS TOXICITY A THESIS SUBMITTED TO THE UNIVERSITE DE TECHNOLOGIE DE COMPIEGNE SORBONNE UNIVERSITES LABORATOIRE DE BIO-MECANIQUE ET BIOINGENIERIE UMR CNRS 7338 – BMBI 18TH OF DECEMBER 2018 For the degree of Doctor Spécialité : Bio-ingénierie et Mathématiques Appliquées Elias ZGHEIB SUPERVISED BY Prof. Frédéric Y. BOIS JURY MEMBERS Mme. Karine AUDOUZE Rapporteur Mr. -
Data Supplement
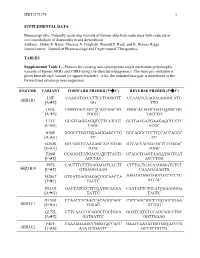
JPET#173179 1 SUPPLEMENTAL DATA Manuscript title: Naturally occurring variants of human aldo-keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin Authors: Onkar S. Bains, Thomas A. Grigliatti, Ronald E. Reid, and K. Wayne Riggs Journal name: Journal of Pharmacology and Experimental Therapeutics TABLES Supplemental Table 1—Primers for creating non-synonymous single nucleotide polymorphic variants of human AKRs and CBR4 using site-directed mutagenesis. The base pair mutation is given beneath each variant [in square brackets]. Also, the mutated base pair is underlined in the forward and reverse primer sequences. ENZYME VARIANT FORWARD PRIMER (5'3') REVERSE PRIMER (5'3') I15F CAAGATGCCCTTCCTGGGGTT CCAACCCCAGGAAGGGCATC AKR1B1 [AT] GG TTG H42L CGGGTACCGCCTCATCGACTG GGGCACAGTCGATGAGGCGG [AT] TGCCC TACCCG L73V GCGTGAGGAGGTCTTCATCGT GCTGACGATGAAGACCTCCTC [CG] CAGC ACGC K90E GGGCCTGGTGGAAGGAGCCTG GGCAGGCTCCTTCCACCAGGC [AG] CC CC G204S GCCAGTCCAAAAGCATCGTGG GTCACCACGATGCTTTTGGAC [GA] TGAC TGGC T288I CCAGGATATGACCATCTTACTC GTAGCTGAGTAAGATGGTCAT [CT] AGCTAC ATCCTGG P87S CACTTTCTTTGAGAGATCCCTT CTTTCCTCACAAGGGATCTCT AKR1B10 [CT] GTGAGGAAAG CAAAGAAAGTG M286T GTGATGAGGAGACGGCAACCA GAGTATGGTTGCCGTCTCCTC [TC] TACTC ATCAC N313D GACTATCCCTTCGATGCAGAA CAATATTCTGCATCGAAGGGA [AG] TATTG TAGTC R170H CCAACTTCAACCACAGGCAGC CTCCAGCTGCCTGTGGTTGAA AKR1C1 [GA] TGGAG GTTGG Q172L CTTCAACCGCAGGCTGCTGGA GGATCATCTCCAGCAGCCTGC [AT] GATGATCC GGTTGAAG F46Y CAATAGAAGCCGGGTACCACC GAATCAATATGGTGGTACCCG AKR1C2 [TA] ATATTTGATTC GCTTCTATTG JPET#173179 -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Functional Role of Pentose Phosphate Pathway and Glutamine in Cancer Cell Metabolism Ibrahim Halil Polat
Functional role of pentose phosphate pathway and glutamine in cancer cell metabolism Ibrahim Halil Polat To cite this version: Ibrahim Halil Polat. Functional role of pentose phosphate pathway and glutamine in cancer cell metabolism. Human health and pathology. Universitat de Barcelona, 2016. English. NNT : 2016GREAS031. tel-01722703 HAL Id: tel-01722703 https://tel.archives-ouvertes.fr/tel-01722703 Submitted on 5 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE LA COMMUNAUTÉ UNIVERSITÉ GRENOBLE ALPES préparée dans le cadre d’une cotutelle entre la Communauté Université Grenoble Alpes et Université de Barcelona Spécialité : BIS-Biotechnologie, instrumentation, signal et imagerie pour la biologie, la médecine et l’environnement Arrêté ministériel : 25 mai 2016 Présentée par « Ibrahim Halil Polat » Thèse dirigée par « Philippe Sabatier » codirigée par « Marta Cascante » préparée au sein des Laboratoires « TIMC-IMAG » et « BQI » dans les Écoles Doctorales « EDISCE » et « Ecole Doctorale de l’Université de Barcelone » Rôle fonctionnel des pentoses phosphates et glutamine dans le métabolisme des cellules cancéreuses Thèse soutenue publiquement le « 4 novembre 2016 », devant le jury composé de : M. Santiago Imperial Rodenas Professeur à l’Université de Barcelone (Président) Mme Loranne Agius Professeur à l’Université de Newcastle Upon Tyne (Rapporteur) M. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

BCAT1 Expression Associates with Ovarian Cancer Progression: Possible Implications in Altered Disease Metabolism
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 31 BCAT1 expression associates with ovarian cancer progression: possible implications in altered disease metabolism Zhi-Qiang Wang1,2, Adnen Faddaoui1,2, Magdalena Bachvarova2, Marie Plante2,3, Jean Gregoire2,3, Marie-Claude Renaud2,3, Alexandra Sebastianelli2,3, Chantal Guillemette4,5, Stéphane Gobeil1,4, Elizabeth Macdonald6, Barbara Vanderhyden6, Dimcho Bachvarov1,2 1Department of Molecular Medicine, Laval University, Québec PQ, Canada 2Centre de recherche du CHU de Québec, L’Hôtel-Dieu de Québec, Québec PQ, Canada 3Department of Obstetrics and Gynecology, Laval University, Québec PQ, Canada 4Centre de recherche du CHU de Québec, CHUL, Québec PQ, Canada 5Faculty of Pharmacy, Laval University, Québec PQ, Canada 6Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, ON, Canada Correspondence to: Dimcho Bachvarov, e-mail: [email protected] Keywords: BCAT1, ovarian cancer, cancer metabolism, metastasis, DNA hypomethylation Received: May 07, 2015 Accepted: August 28, 2015 Published: September 10, 2015 ABSTRACT Previously, we have identified the branched chain amino-acid transaminase 1 (BCAT1) gene as notably hypomethylated in low-malignant potential (LMP) and high-grade (HG) serous epithelial ovarian tumors, compared to normal ovarian tissues. Here we show that BCAT1 is strongly overexpressed in both LMP and HG serous epithelial ovarian tumors, which probably correlates with its hypomethylated status. Knockdown of the BCAT1 expression in epithelial ovarian cancer (EOC) cells led to sharp decrease of cell proliferation, migration and invasion and inhibited cell cycle progression. BCAT1 silencing was associated with the suppression of numerous genes and pathways known previously to be implicated in ovarian tumorigenesis, and the induction of some tumor suppressor genes (TSGs). -

Download Tool
by Submitted in partial satisfaction of the requirements for degree of in in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________ Chair ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2019 by Adolfo Cuesta ii Acknowledgements For me, completing a doctoral dissertation was a huge undertaking that was only possible with the support of many people along the way. First, I would like to thank my PhD advisor, Jack Taunton. He always gave me the space to pursue my own ideas and interests, while providing thoughtful guidance. Nearly every aspect of this project required a technique that was completely new to me. He trusted that I was up to the challenge, supported me throughout, helped me find outside resources when necessary. I remain impressed with his voracious appetite for the literature, and ability to recall some of the most subtle, yet most important details in a paper. Most of all, I am thankful that Jack has always been so generous with his time, both in person, and remotely. I’ve enjoyed our many conversations and hope that they will continue. I’d also like to thank my thesis committee, Kevan Shokat and David Agard for their valuable support, insight, and encouragement throughout this project. My lab mates in the Taunton lab made this such a pleasant experience, even on the days when things weren’t working well. I worked very closely with Tangpo Yang on the mass spectrometry aspects of this project. Xiaobo Wan taught me almost everything I know about protein crystallography. Thank you as well to Geoff Smith, Jordan Carelli, Pat Sharp, Yazmin Carassco, Keely Oltion, Nicole Wenzell, Haoyuan Wang, Steve Sethofer, and Shyam Krishnan, Shawn Ouyang and Qian Zhao. -

Important Roles of the AKR1C2 and SRD5A1 Enzymes in Progesterone
Chemico-Biological Interactions xxx (2014) xxx–xxx Contents lists available at ScienceDirect Chemico-Biological Interactions journal homepage: www.elsevier.com/locate/chembioint Important roles of the AKR1C2 and SRD5A1 enzymes in progesterone metabolism in endometrial cancer model cell lines ⇑ Maša Sinreih a, Maja Anko a, Sven Zukunft b, Jerzy Adamski b,c,d, Tea Lanišnik Rizˇner a, a Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia b Institute of Experimental Genetics, Genome Analysis Centre, Helmholtz Zentrum München, München, Germany c Lehrstuhl für Experimentelle Genetik, Technische Universität München, 85356 Freising-Weihenstephan, Germany d German Centre for Diabetes Research, 85764 Neuherberg, Germany article info abstract Article history: Endometrial cancer is the most frequently diagnosed gynecological malignancy. It is associated with Available online xxxx prolonged exposure to estrogens that is unopposed by progesterone, whereby enhanced metabolism of progesterone may decrease its protective effects, as it can deprive progesterone receptors of their active Keywords: ligand. Furthermore, the 5a-pregnane metabolites formed can stimulate proliferation and may thus 3-Keto/20-keto-reductases contribute to carcinogenesis. The aims of our study were to: (1) identify and quantify progesterone 5a-Reductases metabolites formed in the HEC-1A and Ishikawa model cell lines of endometrial cancer; and (2) pinpoint Pre-receptor metabolism the enzymes involved in progesterone metabolism, and delineate their roles. Progesterone metabolism 5a-Pregnanes studies combined with liquid chromatography–tandem mass spectrometry enabled identification and 4-Pregnenes quantification of the metabolites formed in these cells. Further quantitative PCR analysis and small-inter- fering-RNA-mediated gene silencing identified individual progesterone metabolizing enzymes and their relevant roles.