The University of Chicago the Metaproteomic Analysis Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Comparison of Strand-Specific Transcriptomes Of
Landstorfer et al. BMC Genomics 2014, 15:353 http://www.biomedcentral.com/1471-2164/15/353 RESEARCH ARTICLE Open Access Comparison of strand-specific transcriptomes of enterohemorrhagic Escherichia coli O157:H7 EDL933 (EHEC) under eleven different environmental conditions including radish sprouts and cattle feces Richard Landstorfer1, Svenja Simon2, Steffen Schober3, Daniel Keim2, Siegfried Scherer1 and Klaus Neuhaus1* Abstract Background: Multiple infection sources for enterohemorrhagic Escherichia coli O157:H7 (EHEC) are known, including animal products, fruit and vegetables. The ecology of this pathogen outside its human host is largely unknown and one third of its annotated genes are still hypothetical. To identify genetic determinants expressed under a variety of environmental factors, we applied strand-specific RNA-sequencing, comparing the SOLiD and Illumina systems. Results: Transcriptomes of EHEC were sequenced under 11 different biotic and abiotic conditions: LB medium at pH4, pH7, pH9, or at 15°C; LB with nitrite or trimethoprim-sulfamethoxazole; LB-agar surface, M9 minimal medium, spinach leaf juice, surface of living radish sprouts, and cattle feces. Of 5379 annotated genes in strain EDL933 (genome and plasmid), a surprising minority of only 144 had null sequencing reads under all conditions. We therefore developed a statistical method to distinguish weakly transcribed genes from background transcription. We find that 96% of all genes and 91.5% of the hypothetical genes exhibit a significant transcriptional signal under at least one condition. Comparing SOLiD and Illumina systems, we find a high correlation between both approaches for fold-changes of the induced or repressed genes. The pathogenicity island LEE showed highest transcriptional activity in LB medium, minimal medium, and after treatment with antibiotics. -

The Role of Polyamine Uptake Transporters on Growth and Development of Arabidopsis Thaliana
THE ROLE OF POLYAMINE UPTAKE TRANSPORTERS ON GROWTH AND DEVELOPMENT OF ARABIDOPSIS THALIANA Jigarkumar Patel A Dissertation Submitted to the Graduate College of Bowling Green State University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY May 2015 Committee: Paul Morris, Advisor Wendy D Manning Graduate Faculty Representative Vipaporn Phuntumart Scott Rogers Ray Larsen © 2015 Jigarkumar Patel All Rights Reserved iii ABSTRACT Paul Morris, Advisor Transgenic manipulation of polyamine levels has provided compelling evidence that polyamines enable plants to respond to environmental cues by activation of stress and developmental pathways. Here we show that the chloroplasts of A. thaliana and soybeans contain both an arginine decarboxylase, and an arginase/agmatinase. These two enzymes combine to synthesize putrescine from arginine. Since the sequences of plant arginases show conservation of key residues and the predicted 3D structures of plant agmatinases overlap the crystal structure of the enzyme from Deinococcus radiodurans, we suggest that these enzymes can synthesize putrescine, whenever they have access to the substrate agmatine. Finally, we show that synthesis of putrescine by ornithine decarboxylase takes place in the ER. Thus A. thaliana has two, and soybeans have three separate pathways for the synthesis of putrescine. This study also describes key changes in plant phenotypes in response to altered transport of polyamines. iv Dedicated to my father, Jayantilal Haribhai Patel v ACKNOWLEDGMENTS I would like to thank my advisor, Dr. Paul F. Morris, for helping me learn and grow during my Ph.D. Dr. Morris has an open door policy, and he was always available to answer my questions and provide helpful suggestions. -

RT² Profiler PCR Array (Rotor-Gene® Format) Human Amino Acid Metabolism I
RT² Profiler PCR Array (Rotor-Gene® Format) Human Amino Acid Metabolism I Cat. no. 330231 PAHS-129ZR For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Rotor-Gene Q, other Rotor-Gene cyclers Format R Description The Human Amino Acid Metabolism I RT² Profiler PCR Array profiles the expression of 84 key genes important in biosynthesis and degradation of functional amino acids. Of the 20 amino acids required for protein synthesis, six of them (arginine, cysteine, glutamine, leucine, proline, and tryptophan), collectively known as the functional amino acids, regulate key metabolic pathways involved in cellular growth, and development, as well as other important biological processes such as immunity and reproduction. For example, leucine activates mTOR signaling and increases protein synthesis, leading to lymphocyte proliferation. Therefore, a lack of leucine can compromise immune function. Metabolic pathways interrelated with the biosynthesis and degradation of these amino acids include vitamin and cofactor biosynthesis (such as SAM or S-Adenosyl Methionine) as well as neurotransmitter metabolism (such as glutamate). This array includes genes for mammalian functional amino acid metabolism as well as genes involved in methionine metabolism, important also for nutrient sensing and sulfur metabolism. Using realtime PCR, you can easily and reliably analyze the expression of a focused panel of genes involved in functional amino acid metabolism with this array. For further details, consult the RT² Profiler PCR Array Handbook. Shipping and storage RT² Profiler PCR Arrays in the Rotor-Gene format are shipped at ambient temperature, on dry ice, or blue ice packs depending on destination and accompanying products. -

Enzymatic Synthesis of L-Fucose from L-Fuculose Using a Fucose Isomerase
Kim et al. Biotechnol Biofuels (2019) 12:282 https://doi.org/10.1186/s13068-019-1619-0 Biotechnology for Biofuels RESEARCH Open Access Enzymatic synthesis of L-fucose from L-fuculose using a fucose isomerase from Raoultella sp. and the biochemical and structural analyses of the enzyme In Jung Kim1†, Do Hyoung Kim1†, Ki Hyun Nam1,2 and Kyoung Heon Kim1* Abstract Background: L-Fucose is a rare sugar with potential uses in the pharmaceutical, cosmetic, and food industries. The enzymatic approach using L-fucose isomerase, which interconverts L-fucose and L-fuculose, can be an efcient way of producing L-fucose for industrial applications. Here, we performed biochemical and structural analyses of L-fucose isomerase identifed from a novel species of Raoultella (RdFucI). Results: RdFucI exhibited higher enzymatic activity for L-fuculose than for L-fucose, and the rate for the reverse reaction of converting L-fuculose to L-fucose was higher than that for the forward reaction of converting L-fucose to L-fuculose. In the equilibrium mixture, a much higher proportion of L-fucose (~ ninefold) was achieved at 30 °C and pH 7, indicating that the enzyme-catalyzed reaction favors the formation of L-fucose from L-fuculose. When biochemical analysis was conducted using L-fuculose as the substrate, the optimal conditions for RdFucI activity were determined to be 40 °C and pH 10. However, the equilibrium composition was not afected by reaction temperature in the range 2 of 30 to 50 °C. Furthermore, RdFucI was found to be a metalloenzyme requiring Mn + as a cofactor. The comparative crystal structural analysis of RdFucI revealed the distinct conformation of α7–α8 loop of RdFucI. -
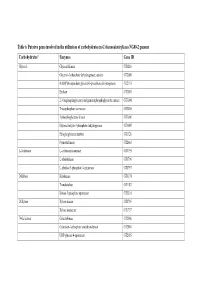
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

SUPPY Liglucosexlmtdh
US 20100314248A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0314248 A1 Worden et al. (43) Pub. Date: Dec. 16, 2010 (54) RENEWABLE BOELECTRONIC INTERFACE Publication Classification FOR ELECTROBOCATALYTC REACTOR (51) Int. Cl. (76) Inventors: Robert M. Worden, Holt, MI (US); C25B II/06 (2006.01) Brian L. Hassler, Lake Orion, MI C25B II/2 (2006.01) (US); Lawrence T. Drzal, Okemos, GOIN 27/327 (2006.01) MI (US); Ilsoon Lee, Okemo s, MI BSD L/04 (2006.01) (US) C25B 9/00 (2006.01) (52) U.S. Cl. ............... 204/403.14; 204/290.11; 204/400; Correspondence Address: 204/290.07; 427/458; 204/252: 977/734; PRICE HENEVELD COOPER DEWITT & LIT 977/742 TON, LLP 695 KENMOOR, S.E., PO BOX 2567 (57) ABSTRACT GRAND RAPIDS, MI 495.01 (US) An inexpensive, easily renewable bioelectronic device useful for bioreactors, biosensors, and biofuel cells includes an elec (21) Appl. No.: 12/766,169 trically conductive carbon electrode and a bioelectronic inter face bonded to a surface of the electrically conductive carbon (22) Filed: Apr. 23, 2010 electrode, wherein the bioelectronic interface includes cata lytically active material that is electrostatically bound directly Related U.S. Application Data or indirectly to the electrically conductive carbon electrode to (60) Provisional application No. 61/172,337, filed on Apr. facilitate easy removal upon a change in pH, thereby allowing 24, 2009. easy regeneration of the bioelectronic interface. 7\ POWER 1 - SUPPY|- LIGLUCOSEXLMtDH?till pi 6.0 - esses&aaaas-exx-xx-xx-xx-xxxxixax-e- Patent Application Publication Dec. 16, 2010 Sheet 1 of 18 US 2010/0314248 A1 Potential (nV) Patent Application Publication Dec. -

Ornithine Decarboxylase Regulates M1 Macrophage Activation And
Ornithine decarboxylase regulates M1 macrophage PNAS PLUS activation and mucosal inflammation via histone modifications Dana M. Hardbowera,b, Mohammad Asimb, Paula B. Luisc, Kshipra Singhb, Daniel P. Barryb, Chunying Yangd,e, Meredith A. Steevese, John L. Clevelandd,e, Claus Schneiderc, M. Blanca Piazuelob, Alain P. Gobertb, and Keith T. Wilsona,b,f,g,h,1 aDepartment of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232; bDivision of Gastroenterology, Hepatology and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; cDepartment of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232; dDepartment of Tumor Biology, Moffitt Cancer Center and Research Institute, Tampa, FL 33612; eDepartment of Cancer Biology, The Scripps Research Institute, Jupiter, FL 33458; fDepartment of Cancer Biology, Vanderbilt University School of Medicine, Nashville, TN 37232; gCenter for Mucosal Inflammation and Cancer, Vanderbilt University Medical Center, Nashville, TN 37232; and hVeterans Affairs Tennessee Valley Healthcare System, Nashville, TN 37232 Edited by Carl F. Nathan, Weill Medical College of Cornell University, New York, NY, and approved December 20, 2016 (received for review September 6, 2016) Macrophage activation is a critical step in host responses during pathogens is macrophage activation. Macrophages have the ca- bacterial infections. Ornithine decarboxylase (ODC), the rate-limiting pacity to alter cytokine/chemokine production and various other enzyme in polyamine metabolism, has been well studied in epithelial functions along the spectrum of M1 or M2 activation based on the cells and is known to have essential roles in many different cellular stimulus detected (12–14). M1 macrophages represent a highly functions. However, its role in regulating macrophage function during proinflammatory and antimicrobial subset of macrophages (12–14). -

WO 2014/207087 Al 31 December 2014 (31.12.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/207087 Al 31 December 2014 (31.12.2014) P O P C T (51) International Patent Classification: Seville (ES). GUTIERREZ GOMEZ, Pablo; Campus C12N 9/00 (2006.01) C12N 9/90 (2006.01) Palmas Altas, Calle Energia Solar 1, E-41014 Seville (ES). C12N 9/02 (2006.01) C12P 5/02 (2006.01) (74) Agent: NEDERLANDSCH OCTROOIBUREAU; J.W. C12N 9/10 (2006.01) C12P 7/76 (2006.01) Frisolaan 13, NL-2517 JS The Hague (NL). C12N 9/12 (2006.01) C12P 7/64 (2006.01) C12N 9/88 (2006.01) C12N 1/1 9 (2006.01) (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (21) International Application Number: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, PCT/EP2014/063484 BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (22) International Filing Date: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, 26 June 2014 (26.06.2014) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (25) Filing Language: English MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (26) Publication Language: English OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (30) Priority Data: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 13382241 .1 26 June 2013 (26.06.2013) EP ZW. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Part 3 Metabolism of Proteins and Nucleic Acids Частина 3 Обмін Білків І Нуклеїнових Кислот
МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ'Я УКРАЇНИ Харківський національний медичний університет PART 3 METABOLISM OF PROTEINS AND NUCLEIC ACIDS Self-Study Guide for Students of General Medicine Faculty in Biochemistry ЧАСТИНА 3 ОБМІН БІЛКІВ І НУКЛЕЇНОВИХ КИСЛОТ Методичні вказівки для підготовки до практичних занять з біологічної хімії (для студентів медичних факультетів) Затверджено вченою радою ХНМУ Протокол № 1 від 26.01.2017 р. Approved by the Scientific Council of KhNMU. Protocol 1 (January 26, 2017) Харків ХНМУ 2017 Metabolism of proteins and nucleic acids : self-study guide for students of general medicine faculty in biochemistry. Part 3: / Comp. : O. Nakonechna, S. Stetsenko, L. Popova, A. Tkachenko. – Kharkiv : KhNMU, 2017. – 56 p. Compilers Nakonechna O. Stetsenko S. Popova L. Tkachenko A. Обмін білків та нуклеїнових кислот : метод. вказ. для підготовки до практ. занять з біологічної хімії (для студ. мед. ф-тів). Ч 3. / упоряд. О.А. Наконечна, С.О. Стеценко, Л.Д. Попова, А.С. Ткаченко. – Харків : ХНМУ, 2017. – 56 с. Упорядники О.А. Наконечна С.О. Стеценко Л.Д. Попова А.С. Ткаченко - 2 - SOURCES For preparing to practical classes in "Biological Chemistry" Basic Sources 1. Біологічна і біоорганічна хімія: у 2 кн.: підруч. Біологічна хімія / Ю.І. Губ- ський, І.В. Ніженковська, М.М. Корда, В.І. Жуков та ін. ; за ред. Ю.І. Губського, І.В. Ніженковської. – Кн. 2. – Київ : ВСВ «Медицина», 2016. – 544 с. 2. Губський Ю.І. Біологічна хімія : підруч. / Ю.І. Губський – Київ– Вінниця: Нова книга, 2007. – 656 с. 3. Губський Ю.І. Біологічна хімія / Губський Ю.І. – Київ–Тернопіль : Укр- медкнига, 2000. – 508 с. 4. Гонський Я.І. -
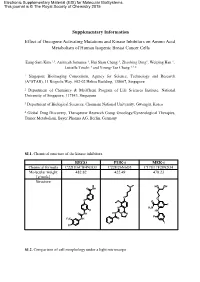
Supplementary Information Effect of Oncogene Activating Mutations And
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2015 Supplementary Information Effect of Oncogene Activating Mutations and Kinase Inhibitors on Amino Acid Metabolism of Human Isogenic Breast Cancer Cells Eung-Sam Kim 1,3, Animesh Samanta 1, Hui Shan Cheng 1, Zhaobing Ding1, Weiping Han 1, Luisella Toschi 4 and Young-Tae Chang 1,2,* 1 Singapore Bioimaging Consortium, Agency for Science, Technology and Research (A*STAR), 11 Biopolis Way, #02-02 Helios Building, 138667, Singapore 2 Department of Chemistry & MedChem Program of Life Sciences Institute, National University of Singapore, 117543, Singapore. 3 Department of Biological Sciences, Chonnam National University, Gwangju, Korea 4 Global Drug Discovery, Therapeutic Research Group Oncology/Gynecological Therapies, Tumor Metabolism, Bayer Pharma AG, Berlin, Germany SI.1. Chemical structure of the kinase inhibitors. REGO PI3K-i MEK-i Chemical formula C22H16ClF4N3O3 C22H26N6O3 C17H17F2IN2O4 Molecular weight 482.82 422.49 478.23 [g/mole] Structure N HO OH O NH O O O O O F N H2N F HN O NH HN N N F F3C NH O N I Cl SI.2. Comparison of cell morphology under a light microscope Scale bar: 100 µm 10x objective lens at Ti microscope (Nikon) SI.3. Scheme of the experimental method for metabolite extraction SI.4. Gradient profile of HPLC run. A : Buffer solution B: Mobile phase H O Time 2 (40 mM NaHPO4, (MeOH/ACN/H2O, Flow rate pH7.8 at RT) 45:45:10, v/v/v) (ml/min) (min) (%) (%) (%) 0 100 0 0 1.0 3 90 10 0 1.0 15 87 13 0 1.0 25 70 30 0 1.0 37 65 35 0 1.0 43 58 42 0 1.0 52 43 57 0 1.0 55 0 100 0 1.0 60 0 0 100 1.0 65 to 80 100 0 0 1.0 SI.5. -

Your Name Here
STRUCTURE ACTIVITY RELATIONSHIPS IN PYRIDOXAL-5’-PHOSPHATE DEPENDENT ENZYMES by SANTIAGO LIMA (Under the Direction of Robert S. Phillips) ABSTRACT Pyridoxal-5’-phosphate (PLP) dependent enzymes are a large and catalytically diverse group of proteins primarily involved in the metabolism of amino acids, amino acid derived compounds, and amino sugars. In this work, three PLP-dependent enzymes, Homo sapiens kynureninase, Pseudomonas dacunhae L-aspartate-β-decarboxylase, and Pyrococcus furiosus tryptophan synthase β-subunit homolog are studied using a variety of biophysical methods. The novel crystal structures for these enzymes are presented, along with the structure of a kynureninase-inhibitor complex, and the analysis of a number of mutants generated to study specific structure activity relationships in them. The results of these analyses reveal the interactions that contribute to substrate specificity in kynureninase, a novel oligomerization scheme and catalytically important residues in aspartate-β-decarboxylase, and the elucidation of the kinetic properties of the P. furiosus tryptophan synthase β-subunit homolog. INDEX WORDS: pyridoxal-5’-phosphate, kynureninase, L-kynurenine hydrolase, 3- hydroxykynurenine, 2HZP, L-aspartate-beta-decarboxylase, aspartate-4- decarboxylase, tryptophan synthase beta subunit homolog, trpb2, tryptophan synthase STRUCTURE ACTIVITY RELATIONSHIPS IN PYRIDOXAL-5’-PHOSPHATE DEPENDENT ENZYMES by SANTIAGO LIMA BS, The University of Georgia, 2001 A Dissertation Submitted to the Graduate Faculty of The University