Crixivan, INN-Indinavir Sulphate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacokinetic Interactions Between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance
life Review Pharmacokinetic Interactions between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance Laura Rombolà 1 , Damiana Scuteri 1,2 , Straface Marilisa 1, Chizuko Watanabe 3, Luigi Antonio Morrone 1, Giacinto Bagetta 1,2,* and Maria Tiziana Corasaniti 4 1 Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, Section of Preclinical and Translational Pharmacology, University of Calabria, 87036 Rende, Italy; [email protected] (L.R.); [email protected] (D.S.); [email protected] (S.M.); [email protected] (L.A.M.) 2 Pharmacotechnology Documentation and Transfer Unit, Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende, Italy 3 Department of Physiology and Anatomy, Tohoku Pharmaceutical University, 981-8558 Sendai, Japan; [email protected] 4 School of Hospital Pharmacy, University “Magna Graecia” of Catanzaro and Department of Health Sciences, University “Magna Graecia” of Catanzaro, 88100 Catanzaro, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-0984-493462 Received: 28 May 2020; Accepted: 30 June 2020; Published: 4 July 2020 Abstract: The therapeutic efficacy of a drug or its unexpected unwanted side effects may depend on the concurrent use of a medicinal plant. In particular, constituents in the medicinal plant extracts may influence drug bioavailability, metabolism and half-life, leading to drug toxicity or failure to obtain a therapeutic response. This narrative review focuses on clinical studies improving knowledge on the ability of selected herbal medicines to influence the pharmacokinetics of co-administered drugs. Moreover, in vitro studies are useful to anticipate potential herbal medicine-drug interactions. -

Indinavir Sulfate Capsule Merck & Co., Inc
CRIXIVAN - indinavir sulfate capsule Merck & Co., Inc. ---------- CRIXIVAN® (INDINAVIR SULFATE) CAPSULES DESCRIPTION CRIXIVAN1 (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 100, 200, 333, and 400 mg of indinavir (corresponding to 125, 250, 416.3, and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin, titanium dioxide, silicon dioxide and sodium lauryl sulfate. The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[[(1,1 dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl)-D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula: Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4• H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol. 1 Registered trademark of MERCK & CO., Inc. COPYRIGHT © 1996, 1997, 1998, 1999, 2004 MERCK & CO., Inc. All rights reserved MICROBIOLOGY Mechanism of Action HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. -
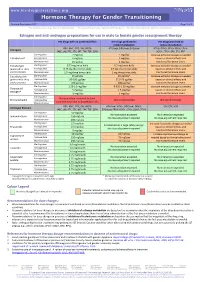
Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 for Personal Use Only
www.hiv-druginteractions.org Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Estrogen and anti-androgen preparations for use in male to female gender reassignment therapy HIV drugs with no predicted effect HIV drugs predicted to HIV drugs predicted to inhibit metabolism induce metabolism RPV, MVC, DTG, RAL, NRTIs ATV/cobi, DRV/cobi, EVG/cobi ATV/r, DRV/r, FPV/r, IDV/r, LPV/r, Estrogens (ABC, ddI, FTC, 3TC, d4T, TAF, TDF, ZDV) SQV/r, TPV/r, EFV, ETV, NVP Starting dose 2 mg/day 1 mg/day Increase estradiol dosage as needed Estradiol oral Average dose 4 mg/day 2 mg/day based on clinical effects and Maximum dose 8 mg/day 4 mg/day monitored hormone levels. Estradiol gel Starting dose 0.75 mg twice daily 0.5 mg twice daily Increase estradiol dosage as needed (preferred for >40 y Average dose 0.75 mg three times daily 0.5 mg three times daily based on clinical effects and and/or smokers) Maximum dose 1.5 mg three times daily 1 mg three times daily monitored hormone levels. Estradiol patch Starting dose 25 µg/day 25 µg/day* Increase estradiol dosage as needed (preferred for >40 y Average dose 50-100 µg/day 37.5-75 µg/day based on clinical effects and and/or smokers) Maximum dose 150 µg/day 100 µg/day monitored hormone levels. Starting dose 1.25-2.5 mg/day 0.625-1.25 mg/day Increase estradiol dosage as needed Conjugated Average dose 5 mg/day 2.5 mg/day based on clinical effects and estrogen† Maximum dose 10 mg/day 5 mg/day monitored hormone levels. -

Indinavir PK Fact Sheet Reviewed March 2016 Page 1 of 2 for Personal Use Only
www.hiv-druginteractions.org Indinavir PK Fact Sheet Reviewed March 2016 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Details Generic Name Indinavir Trade Name Crixivan® Class Protease Inhibitor Molecular Weight 711.88 (as sulphate) Structure HO OH N N H H SO N N 2 4 O HN O H3C CH3 H3C Summary of Key Pharmacokinetic Parameters Plasma half life 1.8 ± 0.4 h Cmax 8.97 ± 2.87 µg/ml (800 mg every 8 hours) Cmin 0.146 µg/ml (800 mg every 8 hours) AUC 21.82 ± 8.11 µg/ml.h (800 mg every 8 hours) Bioavailability Approx 65% Absorption Administration of indinavir with a meal high in calories, fat, and protein resulted in a blunted and reduced absorption with an approximate 80 % reduction in AUC and an 86 % reduction in Cmax. Administration with light meals (e.g., dry toast with jam or fruit conserve, apple juice, and coffee with skimmed or fat–free milk and sugar or corn flakes, skimmed or fat–free milk and sugar) resulted in plasma concentrations comparable to the corresponding fasted values. Protein Binding ~60% Volume of Distribution ~1.74 L/kg [1] CSF:Plasma ratio 0.14 [2] Semen:Plasma ratio 1.9 [2] Renal Clearance <20% as unchanged drug Renal Impairment Safety in patients with impaired renal function has not been studied; less than 20% of indinavir is excreted in the urine as unchanged drug or metabolites. Hepatic Impairment Safety and efficacy of indinavir has not been established in patients with significant underlying liver disorders and should be used with caution. -

Drug-Drug Interaction Between Protease Inhibitors and Statins and Proton Pump Inhibitors
Drug-drug interaction between Protease inhibitors and statins and Proton pump inhibitors Item Type text; Electronic Report Authors Orido, Charles; McKinnon, Samantha Publisher The University of Arizona. Rights Copyright © is held by the author. Download date 01/10/2021 01:48:07 Item License http://rightsstatements.org/vocab/InC/1.0/ Link to Item http://hdl.handle.net/10150/636245 Group 47 :Orido/Samantha 1 Drug-drug interaction between Protease inhibitors and statins and Proton pump inhibitors Course Title: PhPr 862 Date: April 3, 2019 Faculty Advisor: Dr. Dan Malone Students: Charles Orido, Samantha McKinnon Pharm.D. Candidates, Class of 2019 Group 47 :Orido/Samantha 2 Objective The purpose of this article is to provide a systematic review of the pharmacokinetic and clinical data on drug-drug interactions between protease inhibitors (PIs) and statins, atazanavir and proton pump inhibitors (PPIs)and their clinical relevance. Methods A literature search was performed using Medline, EMBASE and google scholar, abstracts from 1970 to 2019 of major conferences were searched and FDA drug information package inserts of the manufacturer of every currently available PI was looked at. All data was summarized and verified by at least two investigators. Results A total of 246 references were identified, 8 of which were studies of pharmacokinetic and pharmacodynamics interactions between simvastatin, lovastatin and protease inhibitors and an additional 7 articles that provided pharmacokinetic of proton pump inhibitors and Atazanavir. Conclusions Protease inhibitors increases the AUC and Cmax of simvastatin by approximately 500% and 517% respectively. Therefore, simvastatin and Lovastatin are not recommended for a co-administration with a protease inhibitor. -

Crixivan (Indinavir Sulfate)
XXXXXXX CRIXIVAN® (INDINAVIR SULFATE) CAPSULES DESCRIPTION CRIXIVAN* (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 100, 200, and 400 mg of indinavir (corresponding to 125, 250, and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin and titanium dioxide. The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H inden-1-yl)-5-[2-[[(1,1-dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2 (phenylmethyl)-D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula: Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4 • H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol. MICROBIOLOGY Mechanism of Action: HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Antiretroviral Activity In Vitro: The in vitro activity of indinavir was assessed in cell lines of lymphoblastic and monocytic origin and in peripheral blood lymphocytes. HIV-1 variants used to infect the different cell types include laboratory-adapted variants, primary clinical isolates and clinical isolates resistant to nucleoside analogue and nonnucleoside inhibitors of the HIV-1 reverse transcriptase. -

The Role of Highly Selective Androgen Receptor (AR) Targeted
P h a s e I I S t u d y o f I t r a c o n a z o l e i n B i o c h e m i c a l R e l a p s e Version 4.0: October 8, 2014 CC# 125513 CC# 125513: Hedgehog Inhibition as a Non-Castrating Approach to Hormone Sensitive Prostate Cancer: A Phase II Study of Itraconazole in Biochemical Relapse Investigational Agent: Itraconazole IND: IND Exempt (IND 116597) Protocol Version: 4.0 Version Date: October 8, 2014 Principal Investigator: Rahul Aggarwal, M.D., HS Assistant Clinical Professor Division of Hematology/Oncology, Department of Medicine University of California San Francisco 1600 Divisadero St. San Francisco, CA94115 [email protected] UCSF Co-Investigators: Charles J. Ryan, M.D., Eric Small, M.D., Professor of Medicine Professor of Medicine and Urology Lawrence Fong, M.D., Terence Friedlander, M.D., Professor in Residence Assistant Clinical Professor Amy Lin, M.D., Associate Clinical Professor Won Kim, M.D., Assistant Clinical Professor Statistician: Li Zhang, Ph.D, Biostatistics Core RevisionHistory October 8, 2014 Version 4.0 November 18, 2013 Version 3.0 January 28, 2013 Version 2.0 July 16, 2012 Version 1.0 Phase II - Itraconazole Page 1 of 79 P h a s e I I S t u d y o f I t r a c o n a z o l e i n B i o c h e m i c a l R e l a p s e Version 4.0: October 8, 2014 CC# 125513 Protocol Signature Page Protocol No.: 122513 Version # and Date: 4.0 - October 8, 2014 1. -

Ongoing Living Update of Potential COVID-19 Therapeutics: Summary of Rapid Systematic Reviews
Ongoing Living Update of Potential COVID-19 Therapeutics: Summary of Rapid Systematic Reviews RAPID REVIEW – July 13th 2020. (The information included in this review reflects the evidence as of the date posted in the document. Updates will be developed according to new available evidence) Disclaimer This document includes the results of a rapid systematic review of current available literature. The information included in this review reflects the evidence as of the date posted in the document. Yet, recognizing that there are numerous ongoing clinical studies, PAHO will periodically update these reviews and corresponding recommendations as new evidence becomes available. 1 Ongoing Living Update of Potential COVID-19 Therapeutics: Summary of Rapid Systematic Reviews Take-home messages thus far: • More than 200 therapeutic options or their combinations are being investigated in more than 1,700 clinical trials. In this review we examined 26 therapeutic options. • Preliminary findings from the RECOVERY Trial showed that low doses of dexamethasone (6 mg of oral or intravenous preparation once daily for 10 days) significantly reduced mortality by one- third in ventilated patients and by one fifth in patients receiving oxygen only. The anticipated RECOVERY Trial findings and WHO’s SOLIDARITY Trial findings both show no benefit via use of hydroxychloroquine and lopinavir/ritonavir in terms of reducing 28-day mortality or reduced time to clinical improvement or reduced adverse events. • Currently, there is no evidence of benefit in critical outcomes (i.e. reduction in mortality) from any therapeutic option (though remdesivir is revealing promise as one option based on 2 randomized controlled trials) and that conclusively allows for safe and effective use to mitigate or eliminate the causative agent of COVID-19. -

ISENTRESS (Raltegravir) for Oral Suspension ------CONTRAINDICATIONS ------Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use --------------------- DOSAGE FORMS AND STRENGTHS --------------------- ISENTRESS safely and effectively. See full prescribing information Film-Coated Tablets: 400 mg (3). for ISENTRESS. Film-Coated Tablets: 600 mg (3). ISENTRESS® (raltegravir) film-coated tablets, for oral use Chewable Tablets: 100 mg scored and 25 mg (3). ® ISENTRESS HD (raltegravir) film-coated tablets, for oral use For Oral Suspension: Single-use packet of 100 mg (3). ISENTRESS® (raltegravir) chewable tablets, for oral use ® ISENTRESS (raltegravir) for oral suspension ------------------------------- CONTRAINDICATIONS ------------------------------- Initial U.S. Approval: 2007 None (4). ----------------------------INDICATIONS AND USAGE ---------------------------- ----------------------- WARNINGS AND PRECAUTIONS ----------------------- Adult Patients: Severe, potentially life-threatening and fatal skin reactions have been ISENTRESS and ISENTRESS HD are human immunodeficiency virus reported. This includes cases of Stevens-Johnson syndrome, integrase strand transfer inhibitors (HIV-1 INSTI) indicated in hypersensitivity reaction and toxic epidermal necrolysis. Immediately combination with other antiretroviral agents for the treatment of HIV-1 discontinue treatment with ISENTRESS or ISENTRESS HD and infection in adult patients (1). other suspect agents if severe hypersensitivity, severe rash, or rash Pediatric Patients: with systemic symptoms or liver aminotransferase -

Clinically Relevant Drug Interactions with Antiepileptic Drugs
British Journal of Clinical Pharmacology DOI:10.1111/j.1365-2125.2005.02529.x Clinically relevant drug interactions with antiepileptic drugs Emilio Perucca Institute of Neurology IRCCS C. Mondino Foundation, Pavia, and Clinical Pharmacology Unit, Department of Internal Medicine and Therapeutics, University of Pavia, Pavia, Italy Correspondence Some patients with difficult-to-treat epilepsy benefit from combination therapy with Emilio Perucca MD, PhD, Clinical two or more antiepileptic drugs (AEDs). Additionally, virtually all epilepsy patients will Pharmacology Unit, Department of receive, at some time in their lives, other medications for the management of Internal Medicine and Therapeutics, associated conditions. In these situations, clinically important drug interactions may University of Pavia, Piazza Botta 10, occur. Carbamazepine, phenytoin, phenobarbital and primidone induce many cyto- 27100 Pavia, Italy. chrome P450 (CYP) and glucuronyl transferase (GT) enzymes, and can reduce Tel: + 390 3 8298 6360 drastically the serum concentration of associated drugs which are substrates of the Fax: + 390 3 8222 741 same enzymes. Examples of agents whose serum levels are decreased markedly by E-mail: [email protected] enzyme-inducing AEDs, include lamotrigine, tiagabine, several steroidal drugs, cyclosporin A, oral anticoagulants and many cardiovascular, antineoplastic and psy- chotropic drugs. Valproic acid is not enzyme inducer, but it may cause clinically relevant drug interactions by inhibiting the metabolism of selected substrates, most Keywords notably phenobarbital and lamotrigine. Compared with older generation agents, most antiepileptic drugs, drug interactions, of the recently developed AEDs are less likely to induce or inhibit the activity of CYP enzyme induction, enzyme inhibition, or GT enzymes. However, they may be a target for metabolically mediated drug epilepsy, review interactions, and oxcarbazepine, lamotrigine, felbamate and, at high dosages, topira- mate may stimulate the metabolism of oral contraceptive steroids. -

Proposal to Waive in Vivo Bioequivalence Requirements for the Who Model List of Essential Medicines Immediate Release, Solid Oral Dosage Forms
Working document QAS/04.109/Rev.1 page 1 WORLD HEALTH ORGANIZATION ORGANISATION MONDIALE DE LA SANTE PROPOSAL TO WAIVE IN VIVO BIOEQUIVALENCE REQUIREMENTS FOR THE WHO MODEL LIST OF ESSENTIAL MEDICINES IMMEDIATE RELEASE, SOLID ORAL DOSAGE FORMS This document has been revised by Professor Jennifer B. Dressman, Institut für Pharmazeutische Technologie, Biozentrum, Johann Wolfgang Goethe-Universität, Frankfurt/Main, Germany. It has followed the steps given in the schedule on page 2 herein. It has been very widely distributed and numerous comments have been incorporated. Please address any comments and/or corrections you may have to Dr S. Kopp, Quality Assurance and Safety: Medicines, Medicines Policy and Standards, World Health Organization, 1211 Geneva 27, Switzerland, fax: (+41 22) 791 4730 or e-mail: [email protected] (with a copy to [email protected]) by 20 October 2005. © World Health Organization 2005 All rights reserved. This draft is intended for a restricted audience only, i.e. the individuals and organizations having received this draft. The draft may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in any form or by any means outside these individuals and organizations (including the organizations’ concerned staff and member organizations) without the permission of WHO. The draft should not be displayed on any website. Please send any request for permission to: Dr Sabine Kopp, Quality Assurance & Safety: Medicines (QSM), Department of Medicines Policy and Standards (PSM), World Health Organization, CH-1211 Geneva 27, Switzerland. Fax: (41-22) 791 4730; e-mails: [email protected]; [email protected] The designations employed and the presentation of the material in this draft do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Crixivan® (Indinavir Sulfate) Capsules
CRIXIVAN® (INDINAVIR SULFATE) CAPSULES DESCRIPTION CRIXIVAN® (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 200 and 400 mg of indinavir (corresponding to 250 and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin and titanium dioxide. The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H- inden-1-yl)-5-[2-[[(1,1-dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl)- D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula: Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4 • H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol. MICROBIOLOGY Mechanism of Action: HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Antiretroviral Activity In Vitro: The in vitro activity of indinavir was assessed in cell lines of lymphoblastic and monocytic origin and in peripheral blood lymphocytes. HIV-1 variants used to infect the different cell types include laboratory-adapted variants, primary clinical isolates and clinical isolates resistant to nucleoside analogue and nonnucleoside inhibitors of the HIV-1 reverse transcriptase.