The Mechanism of Drug Nephrotoxicity and the Methods for Preventing Kidney Damage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Highlights of Prescribing Information
HIGHLIGHTS OF PRESCRIBING INFORMATION --------------------------WARNINGS AND PRECAUTIONS-------------------- These highlights do not include all the information needed to use • New onset or worsening renal impairment: Can include acute VIREAD safely and effectively. See full prescribing information renal failure and Fanconi syndrome. Assess creatinine clearance for VIREAD. (CrCl) before initiating treatment with VIREAD. Monitor CrCl and ® serum phosphorus in patients at risk. Avoid administering VIREAD (tenofovir disoproxil fumarate) tablets, for oral use VIREAD with concurrent or recent use of nephrotoxic drugs. (5.3) VIREAD® (tenofovir disoproxil fumarate) powder, for oral use • Coadministration with Other Products: Do not use with other Initial U.S. Approval: 2001 tenofovir-containing products (e.g., ATRIPLA, COMPLERA, and TRUVADA). Do not administer in combination with HEPSERA. WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH (5.4) STEATOSIS and POST TREATMENT EXACERBATION OF HEPATITIS • HIV testing: HIV antibody testing should be offered to all HBV- infected patients before initiating therapy with VIREAD. VIREAD See full prescribing information for complete boxed warning. should only be used as part of an appropriate antiretroviral • Lactic acidosis and severe hepatomegaly with steatosis, combination regimen in HIV-infected patients with or without HBV including fatal cases, have been reported with the use of coinfection. (5.5) nucleoside analogs, including VIREAD. (5.1) • Decreases in bone mineral density (BMD): Consider assessment • Severe acute exacerbations of hepatitis have been reported of BMD in patients with a history of pathologic fracture or other in HBV-infected patients who have discontinued anti- risk factors for osteoporosis or bone loss. (5.6) hepatitis B therapy, including VIREAD. Hepatic function • Redistribution/accumulation of body fat: Observed in HIV-infected should be monitored closely in these patients. -

Pharmacokinetic Interactions Between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance
life Review Pharmacokinetic Interactions between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance Laura Rombolà 1 , Damiana Scuteri 1,2 , Straface Marilisa 1, Chizuko Watanabe 3, Luigi Antonio Morrone 1, Giacinto Bagetta 1,2,* and Maria Tiziana Corasaniti 4 1 Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, Section of Preclinical and Translational Pharmacology, University of Calabria, 87036 Rende, Italy; [email protected] (L.R.); [email protected] (D.S.); [email protected] (S.M.); [email protected] (L.A.M.) 2 Pharmacotechnology Documentation and Transfer Unit, Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende, Italy 3 Department of Physiology and Anatomy, Tohoku Pharmaceutical University, 981-8558 Sendai, Japan; [email protected] 4 School of Hospital Pharmacy, University “Magna Graecia” of Catanzaro and Department of Health Sciences, University “Magna Graecia” of Catanzaro, 88100 Catanzaro, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-0984-493462 Received: 28 May 2020; Accepted: 30 June 2020; Published: 4 July 2020 Abstract: The therapeutic efficacy of a drug or its unexpected unwanted side effects may depend on the concurrent use of a medicinal plant. In particular, constituents in the medicinal plant extracts may influence drug bioavailability, metabolism and half-life, leading to drug toxicity or failure to obtain a therapeutic response. This narrative review focuses on clinical studies improving knowledge on the ability of selected herbal medicines to influence the pharmacokinetics of co-administered drugs. Moreover, in vitro studies are useful to anticipate potential herbal medicine-drug interactions. -

Indinavir Sulfate Capsule Merck & Co., Inc
CRIXIVAN - indinavir sulfate capsule Merck & Co., Inc. ---------- CRIXIVAN® (INDINAVIR SULFATE) CAPSULES DESCRIPTION CRIXIVAN1 (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 100, 200, 333, and 400 mg of indinavir (corresponding to 125, 250, 416.3, and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin, titanium dioxide, silicon dioxide and sodium lauryl sulfate. The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[[(1,1 dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl)-D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula: Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4• H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol. 1 Registered trademark of MERCK & CO., Inc. COPYRIGHT © 1996, 1997, 1998, 1999, 2004 MERCK & CO., Inc. All rights reserved MICROBIOLOGY Mechanism of Action HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. -

Focal Segmental Glomerulosclerosis in Systemic Lupus Erythematosus
Relato de Caso Focal Segmental Glomerulosclerosis in Systemic Lupus Erythematosus: One or Two Glomerular Diseases? Glomerulosclerose Segmentar e Focal em Lúpus Eritematoso Sistêmico: Uma ou Duas Doenças? Rogério Barbosa de Deus1, Luis Antonio Moura1, Marcello Fabiano Franco2, Gianna Mastroianni Kirsztajn1 1 Nephrology Division and 2 Department of Pathology - Universidade Federal de São Paulo – EPM/UNIFESP- São Paulo. ABSTRACT Some patients with clinical and/or laboratory diagnosis of systemic lupus erythematosus (SLE) present with nephritis which from the morphological point of view does not fit in one of the 6 classes described in the WHO classification of lupus nephritis. On the other hand, nonlupus nephritis in patients with confirmed SLE is rarely reported. This condition may not be so uncommon as it seems. The associated glomerular lesions most frequently described are amyloidosis and focal segmental glomerulosclerosis (FSGS). We report on a 46 year-old, caucasian woman, who fulfilled the American College of Rheumatology criteria for SLE diagnosis: arthritis, positive anti-DNA, ANA, anti-Sm antibodies, and cutaneous maculae. During the follow-up, she presented arthralgias, alopecia, vasculitis, lower extremities edema and decreased serum levels of C3 and C4. Proteinuria was initially nephrotic, but reached negative levels. The serum creatinine varied from 0.7 to 3.0 mg/dl. The patient was submitted to the first renal biopsy at admission and to the second one, 3 years later, with diagnosis of minimal change disease and FSGS, respectively. No deposits were demonstrated by immunofluorescence. In the present case, we believe that the patient had SLE and developed an idiopathic disease of the minimal change disease-FSGS spectrum. -

Novel Therapeutics for Epstein–Barr Virus
molecules Review Novel Therapeutics for Epstein–Barr Virus Graciela Andrei *, Erika Trompet and Robert Snoeck Laboratory of Virology and Chemotherapy, Department of Microbiology and Immunology, Rega Institute for Medical Research, KU Leuven, 3000 Leuven, Belgium; [email protected] (E.T.); [email protected] (R.S.) * Correspondence: [email protected]; Tel.: +32-16-321-915 Academic Editor: Stefano Aquaro Received: 15 February 2019; Accepted: 4 March 2019; Published: 12 March 2019 Abstract: Epstein–Barr virus (EBV) is a human γ-herpesvirus that infects up to 95% of the adult population. Primary EBV infection usually occurs during childhood and is generally asymptomatic, though the virus can cause infectious mononucleosis in 35–50% of the cases when infection occurs later in life. EBV infects mainly B-cells and epithelial cells, establishing latency in resting memory B-cells and possibly also in epithelial cells. EBV is recognized as an oncogenic virus but in immunocompetent hosts, EBV reactivation is controlled by the immune response preventing transformation in vivo. Under immunosuppression, regardless of the cause, the immune system can lose control of EBV replication, which may result in the appearance of neoplasms. The primary malignancies related to EBV are B-cell lymphomas and nasopharyngeal carcinoma, which reflects the primary cell targets of viral infection in vivo. Although a number of antivirals were proven to inhibit EBV replication in vitro, they had limited success in the clinic and to date no antiviral drug has been approved for the treatment of EBV infections. We review here the antiviral drugs that have been evaluated in the clinic to treat EBV infections and discuss novel molecules with anti-EBV activity under investigation as well as new strategies to treat EBV-related diseases. -
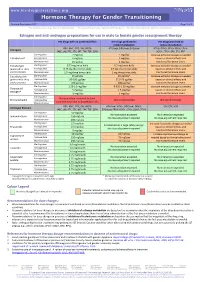
Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 for Personal Use Only
www.hiv-druginteractions.org Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Estrogen and anti-androgen preparations for use in male to female gender reassignment therapy HIV drugs with no predicted effect HIV drugs predicted to HIV drugs predicted to inhibit metabolism induce metabolism RPV, MVC, DTG, RAL, NRTIs ATV/cobi, DRV/cobi, EVG/cobi ATV/r, DRV/r, FPV/r, IDV/r, LPV/r, Estrogens (ABC, ddI, FTC, 3TC, d4T, TAF, TDF, ZDV) SQV/r, TPV/r, EFV, ETV, NVP Starting dose 2 mg/day 1 mg/day Increase estradiol dosage as needed Estradiol oral Average dose 4 mg/day 2 mg/day based on clinical effects and Maximum dose 8 mg/day 4 mg/day monitored hormone levels. Estradiol gel Starting dose 0.75 mg twice daily 0.5 mg twice daily Increase estradiol dosage as needed (preferred for >40 y Average dose 0.75 mg three times daily 0.5 mg three times daily based on clinical effects and and/or smokers) Maximum dose 1.5 mg three times daily 1 mg three times daily monitored hormone levels. Estradiol patch Starting dose 25 µg/day 25 µg/day* Increase estradiol dosage as needed (preferred for >40 y Average dose 50-100 µg/day 37.5-75 µg/day based on clinical effects and and/or smokers) Maximum dose 150 µg/day 100 µg/day monitored hormone levels. Starting dose 1.25-2.5 mg/day 0.625-1.25 mg/day Increase estradiol dosage as needed Conjugated Average dose 5 mg/day 2.5 mg/day based on clinical effects and estrogen† Maximum dose 10 mg/day 5 mg/day monitored hormone levels. -

Treatment of Warts with Topical Cidofovir in a Pediatric Patient
Volume 25 Number 5| May 2019| Dermatology Online Journal || Case Report 25(5):6 Treatment of warts with topical cidofovir in a pediatric patient Melissa A Nickles BA, Artem Sergeyenko MD, Michelle Bain MD Affiliations: Department of Dermatology, University of Illinois at Chicago College of Medicine, Chicago, llinois, USA Corresponding Author: Artem Sergeyenko MD, 808 South Wood Street Suite 380, Chicago, IL 60612, Tel: 847-338-0037, Email: a.serge04@gmail topical cidofovir is effective in treating HPV lesions Abstract and molluscum contagiosum in adult patients with Cidofovir is an antiviral nucleotide analogue with HIV/AIDS [2]. Case reports have also found topical relatively new treatment capacities for cidofovir to effectively treat anogenital squamous dermatological conditions, specifically verruca cell carcinoma (SCC), bowenoid papulosis, vulgaris caused by human papilloma virus infection. condyloma acuminatum, Kaposi sarcoma, and HSV-II In a 10-year old boy with severe verruca vulgaris in adult patients with HIV/AIDS [3]. Cidofovir has recalcitrant to multiple therapies, topical 1% experimentally been shown to be effective in cidofovir applied daily for eight weeks proved to be an effective treatment with no adverse side effects. treating genital condyloma acuminata in adult This case report, in conjunction with multiple immunocompetent patients [4] and in a pediatric published reports, suggests that topical 1% cidofovir case [5]. is a safe and effective treatment for viral warts in Cidofovir has also been used in pediatric patients to pediatric patients. cure verruca vulgaris recalcitrant to traditional treatment therapies. There have been several reports Keywords: cidofovir, verruca vulgaris, human papilloma that topical 1-3% cidofovir cream applied once or virus twice daily is effective in treating verruca vulgaris with no systemic side effects and low rates of recurrence in immunocompetent children [6-8], as Introduction well as in immunocompromised children [9, 10]. -

Effect of Renin-Angiotensin System Inhibitors on the Comparative Nephrotoxicity of Nsaids and Opioids During Hospitalization
Kidney360 Publish Ahead of Print, published on April 27, 2020 as doi:10.34067/KID.0001432020 Effect of renin-angiotensin system inhibitors on the comparative nephrotoxicity of NSAIDs and opioids during hospitalization Todd A. Miano1,2,3; Michael G. S. Shashaty2,4; Wei Yang1,2,3; Jeremiah R. Brown5,6,7; Athena Zuppa8; Sean Hennessy1,2,3 Affiliations: 1. Center for Pharmacoepidemiology Research and Training, Perelman School of Medicine at the University of Pennsylvania 2. Center for Clinical Epidemiology and Biostatistics, Perelman School of Medicine at the University of Pennsylvania 3. Department of Biostatistics, Epidemiology, and Informatics, Perelman School of Medicine at the University of Pennsylvania 4. Pulmonary, Allergy, and Critical Care Division, Perelman School of Medicine at the University of Pennsylvania 5. The Dartmouth Institute for Health Policy and Clinical Practice, Lebanon, New Hampshire 6. Department of Epidemiology, Geisel School of Medicine, Hanover, New Hampshire 7. Department of Biomedical Data Science, Geisel School of Medicine, Hanover, New Hampshire 8. Department of Anesthesiology and Critical Care Medicine, The Children's Hospital of Philadelphia, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA Corresponding author: Todd A. Miano, PharmD, PhD; Address: 423 Guardian Drive, 725 Blockley Hall, Philadelphia, PA 19104; E-mail: [email protected] ; twitter: @Miano81 Copyright 2020 by American Society of Nephrology. Abstract Background: Nonsteroidal anti-inflammatory drugs (NSAIDS) are increasingly important alternatives to opioids for analgesia during hospitalization, as health systems implement opioid minimization initiatives. Increasing NSAID use may increase acute kidney injury (AKI) rates, particularly in patients with predisposing risk factors. Inconclusive data in outpatient populations suggests that NSAID nephrotoxicity is magnified by renin-angiotensin system inhibitors (RAS-I). -

Annex I Summary of Product Characteristics
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 4 1. NAME OF THE MEDICINAL PRODUCT VISTIDE 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each vial contains cidofovir equivalent to 375 mg/5 ml (75 mg/ml) cidofovir anhydrous. The formulation is adjusted to pH 7.4. 3. PHARMACEUTICAL FORM Concentrate for solution for infusion 4. CLINICAL PARTICULARS 4.1 Therapeutic Indication Cidofovir is indicated for the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS) and without renal dysfunction. Until further experience is gained, cidofovir should be used only when other agents are considered unsuitable. 4.2 Posology and Method of Administration Before each administration of cidofovir, serum creatinine and urine protein levels should be investigated. The recommended dosage, frequency, or infusion rate must not be exceeded. Cidofovir must be diluted in 100 milliliters 0.9% (normal) saline prior to administration. To minimise potential nephrotoxicity, oral probenecid and intravenous saline prehydration must be administered with each cidofovir infusion. Dosage in Adults • Induction Treatment. The recommended dose of cidofovir is 5 mg/kg body weight (given as an intravenous infusion at a constant rate over 1 hr) administered once weekly for two consecutive weeks. • Maintenance Treatment. Beginning two weeks after the completion of induction treatment, the recommended maintenance dose of cidofovir is 5 mg/kg body weight (given as an intravenous infusion at a constant rate over 1 hr) administered once every two weeks. Cidofovir therapy should be discontinued and intravenous hydration is advised if serum creatinine increases by = 44 µmol/L (= 0.5 mg/dl), or if persistent proteinuria = 2+ develops. • Probenecid. -

Non-Steroidal Anti-Inflammatory Nephrotoxicity* RICHARD MANIGLIA, B.A.T ALLAN B
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 18, No. 3 Copyright © 1988, Institute for Clinical Science, Inc. Non-Steroidal Anti-Inflammatory Nephrotoxicity* RICHARD MANIGLIA, B.A.t ALLAN B. SCHWARTZ, M.D.,$ and SHEILA MORIBER-KATZ, M.D.f§ f Department of Pathology and Laboratory Medicine, tDepartment of Medicine, §Division of Renal Pathology and Electron Microscopy, Hahnemann University School of Medicine, Philadelphia, PA 19102 ABSTRACT Non-steroidal anti-inflammatory drugs have a wide range of use in clini cal practice because of their analgesic and anti-inflammatory properties. However, their potential nephrotoxicity has been noted. The case histo ries were studied, retrospectively, in 13 patients who were taking non-ste- roidal anti-inflammatory drugs as follows: four on fenoprofen (Nalfon®), three on naproxen (Naprosyn®), two on ibuprofen (Motrin®), two on sulin- dac (Clinoril®), one on tolmetin (Tolectin®), and one on indomethacin (Indocin®) and who exhibited abnormal urinalysis or a deterioration in renal function. Nine of the patients underwent renal biopsies, and eight of these biopsies were positive for interstitial nephritis. In addition to the presentation of additional cases of non-steroidal anti-inflammatory drug nephrotoxicity, a brief review of the current theories of the nephrotoxic mechanism is presented. Introduction this paper will be the non-aspirin, non steroidal anti-inflammatory drugs Numerous reports exist that document (NSAID). Since these drugs have a wide the nephrotoxicity of drugs such as the spectrum of use in clinical practice, their synthetic penicillin (methacillin, carben- deleterious actions are of more than just icillin, ampicillin), other antibiotics academic interest. It is hoped that by (aminoglycosides, amphotericin B), understanding the nephrotoxic patho thiazide and loop diuretics, and anti physiologic mechanisms of non-steroidal neoplastic agents. -

What Is Lupus Nephritis
What is lupus nephritis (LN)? Lupus nephritis (LN) is inflammation of the kidney that occurs as a common symptom of systemic lupus erythematosus (SLE), also known as lupus. Proteins in the immune system called antibodies damage important structures in the kidney. ⅱ Why are the kidneys important? To understand how lupus nephritis damages the kidney, it is important to understand how the kidneys work. The kidneys’ main function is to filter out excess waste and water from the blood through the urine. Kidneys also balance the salts and minerals circulating in the blood, help control blood pressure and make red blood cells. So, when the kidneys are damaged or fail, they can’t do their job as well. As a result, the kidneys are not able to filter out waste and water into the urine causing it to stay in the blood. What are the signs and symptoms of LN? Signs to ask the doctor about include blood in the urine or foamy urine which can mean that there is excess protein. Other signs to notice are swelling of legs, ankles, hands or tissue around the eyes as well as weight gain that can be caused by fluid the body isn’t getting rid of. ⅲ Symptoms of lupus nephritis also include high blood pressure, joint/muscle pain, high levels of waste (creatinine) in the blood, or impaired/failing kidney. ⅳ How common is LN? Lupus nephritis is the most common complication of lupus. Five out of 10 adults with lupus will have lupus nephritis, while eight out of 10 children with lupus will have kidney damage, which usually stems from lupus nephritis. -

Pathophysiologic Mechanisms of Selected Types of Nephrotoxicity 3/12/14, 10:45 AM
Pathophysiologic Mechanisms of Selected Types of Nephrotoxicity 3/12/14, 10:45 AM Today News Reference Education Log In Register Pathophysiologic Mechanisms of Selected Types of Nephrotoxicity Author: Piper Julie Hughes, MD, MS; Chief Editor: Vecihi Batuman, MD, FACP, FASN more... Updated: Dec 18, 2013 Aminoglycoside Nephrotoxicity Aminoglycosides preferentially affect the proximal tubular cells. These agents are freely filtered by the glomeruli and quickly taken up by the proximal tubular epithelial cells, where they are incorporated into lysosomes after first interacting with phospholipids on the brush border membranes. They exert their main toxic effect within the tubular cell by altering phospholipid metabolism. In addition to their direct effect on cells, aminoglycosides cause renal vasoconstriction. The 2 critical factors in the development of acute kidney injury (AKI) secondary to aminoglycoside nephrotoxicity are dosing and duration of therapy. Aminoglycoside uptake by the tubules is a saturable phenomenon, so uptake is limited after a single dose. Thus, a single daily large dose is preferable to 3 doses per day. One dose per day presumably causes less accumulation in the tubular cells once the saturation point is reached.[1, 2] For further information, go to Acute Renal Failure and Acute Tubular Necrosis. Amphotericin B Nephrotoxicity Amphotericin B binds to sterols in cell membranes, thereby creating pores that compromise membrane integrity and increase membrane permeability. It binds not only to ergosterol in fungal cell walls but also to cholesterol in human cell membranes; this is what accounts for its nephrotoxicity. Characteristic electrolyte abnormalities include wasting of potassium and magnesium secondary to increased permeability of the cell membranes.