Crixivan® (Indinavir Sulfate) Capsules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

35 Cyproterone Acetate and Ethinyl Estradiol Tablets 2 Mg/0
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrCYESTRA®-35 cyproterone acetate and ethinyl estradiol tablets 2 mg/0.035 mg THERAPEUTIC CLASSIFICATION Acne Therapy Paladin Labs Inc. Date of Preparation: 100 Alexis Nihon Blvd, Suite 600 January 17, 2019 St-Laurent, Quebec H4M 2P2 Version: 6.0 Control # 223341 _____________________________________________________________________________________________ CYESTRA-35 Product Monograph Page 1 of 48 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION ....................................................................... 3 SUMMARY PRODUCT INFORMATION ............................................................................................. 3 INDICATION AND CLINICAL USE ..................................................................................................... 3 CONTRAINDICATIONS ........................................................................................................................ 3 WARNINGS AND PRECAUTIONS ....................................................................................................... 4 ADVERSE REACTIONS ....................................................................................................................... 13 DRUG INTERACTIONS ....................................................................................................................... 16 DOSAGE AND ADMINISTRATION ................................................................................................ 20 OVERDOSAGE .................................................................................................................................... -

Pharmacokinetic Interactions Between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance
life Review Pharmacokinetic Interactions between Herbal Medicines and Drugs: Their Mechanisms and Clinical Relevance Laura Rombolà 1 , Damiana Scuteri 1,2 , Straface Marilisa 1, Chizuko Watanabe 3, Luigi Antonio Morrone 1, Giacinto Bagetta 1,2,* and Maria Tiziana Corasaniti 4 1 Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, Section of Preclinical and Translational Pharmacology, University of Calabria, 87036 Rende, Italy; [email protected] (L.R.); [email protected] (D.S.); [email protected] (S.M.); [email protected] (L.A.M.) 2 Pharmacotechnology Documentation and Transfer Unit, Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende, Italy 3 Department of Physiology and Anatomy, Tohoku Pharmaceutical University, 981-8558 Sendai, Japan; [email protected] 4 School of Hospital Pharmacy, University “Magna Graecia” of Catanzaro and Department of Health Sciences, University “Magna Graecia” of Catanzaro, 88100 Catanzaro, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-0984-493462 Received: 28 May 2020; Accepted: 30 June 2020; Published: 4 July 2020 Abstract: The therapeutic efficacy of a drug or its unexpected unwanted side effects may depend on the concurrent use of a medicinal plant. In particular, constituents in the medicinal plant extracts may influence drug bioavailability, metabolism and half-life, leading to drug toxicity or failure to obtain a therapeutic response. This narrative review focuses on clinical studies improving knowledge on the ability of selected herbal medicines to influence the pharmacokinetics of co-administered drugs. Moreover, in vitro studies are useful to anticipate potential herbal medicine-drug interactions. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

The Differential Effects of Statins on the Metastatic Behaviour of Prostate Cancer
British Journal of Cancer (2012) 106, 1689–1696 & 2012 Cancer Research UK All rights reserved 0007 – 0920/12 www.bjcancer.com The differential effects of statins on the metastatic behaviour of prostate cancer *,1 1 1 2,3 2,3 2,4 1,2,4 M Brown , C Hart , T Tawadros , V Ramani , V Sangar , M Lau and N Clarke 1 Genito Urinary Cancer Research Group, University of Manchester, Paterson Institute for Cancer Research, Manchester Academic Health Science Centre, 2 The Christie NHS Foundation Trust, Wilmslow Road, Withington, Manchester M20 4BX, UK; Department of Urology, The Christie NHS Foundation 3 Trust, Wilmslow Road, Manchester M20 4BX, UK; Department of Urology, University Hospital of South Manchester NHS Trust, Manchester M23 9LT, 4 UK; Department of Urology, Salford Royal NHS Foundation Trust, Stott Lane, Manchester M6 8HD, UK BACKGROUND: Although statins do not affect the incidence of prostate cancer (CaP), usage reduces the risk of clinical progression and mortality. Although statins are known to downregulate the mevalonate pathway, the mechanism by which statins reduce CaP progression is unknown. METHODS: Bone marrow stroma (BMS) was isolated with ethical approval from consenting patients undergoing surgery for non- malignant disease. PC-3 binding, invasion and colony formation within BMS was assessed by standardised in vitro co-culture assays in the presence of different statins. RESULTS: Statins act directly on PC-3 cells with atorvastatin, mevastatin, simvastatin (1 mM) and rosuvastatin (5 mM), but not pravastatin, significantly reducing invasion towards BMS by an average of 66.68% (range 53.93–77.04%; Po0.05) and significantly reducing both 2 2 number (76.2±8.29 vs 122.9±2.48; P ¼ 0.0055) and size (0.2±0.0058 mm vs 0.27±0.012 mm ; P ¼ 0.0019) of colonies formed within BMS. -

What Precautions Should We Use with Statins for Women of Childbearing
CLINICAL INQUIRIES What precautions should we use with statins for women of childbearing age? Chaitany Patel, MD, Lisa Edgerton, PharmD New Hanover Regional Medical Center, Wilmington, North Carolina Donna Flake, MSLS, MSAS Coastal Area Health Education Center, Wilmington, NC EVIDENCE- BASED ANSWER Statins are contraindicated for women who are on its low tissue-penetration properties. pregnant or breastfeeding. Data evaluating statin Cholesterol-lowering with simvastatin 40 mg/d did use for women of childbearing age is limited; how- not disrupt menstrual cycles or effect luteal phase ever, they may be used cautiously with adequate duration (strength of recommendation: C). contraception. Pravastatin may be preferred based CLINICAL COMMENTARY Use statins only as a last resort Before reading this review, I had not been for women of childbearing age ® Dowdenaware Health of the serious Media effects of statin medications I try to follow the USPSTF recommendations and on the developing fetus. In conversations with not screen women aged <45 years without coro- my colleagues, I found that the adverse effects nary artery disease riskCopyright factors for Fhyperlipidemia.or personalof usestatins onlyduring pregnancy are not readily When a woman of any age needs treatment, my known. Such information needs to be more first-line therapy is lifestyle modification. Given the widely disseminated. risks of statin drugs to the developing fetus, Ariel Smits, MD women with childbearing potential should give Department of Family Medicine, Oregon Health & Science fully informed consent and be offered reliable University, Portland contraception before stating statin therapy. I Evidence summary anal, cardiac, tracheal, esophageal, renal, Hydroxymethyl glutaryl coenzyme A and limb deficiency (VACTERL associa- (HMG CoA) reductase inhibitors, com- tion), intrauterine growth retardation monly called statins, have been on the (IUGR), and demise in fetuses exposed market since the late 1980s. -

Indinavir Sulfate Capsule Merck & Co., Inc
CRIXIVAN - indinavir sulfate capsule Merck & Co., Inc. ---------- CRIXIVAN® (INDINAVIR SULFATE) CAPSULES DESCRIPTION CRIXIVAN1 (indinavir sulfate) is an inhibitor of the human immunodeficiency virus (HIV) protease. CRIXIVAN Capsules are formulated as a sulfate salt and are available for oral administration in strengths of 100, 200, 333, and 400 mg of indinavir (corresponding to 125, 250, 416.3, and 500 mg indinavir sulfate, respectively). Each capsule also contains the inactive ingredients anhydrous lactose and magnesium stearate. The capsule shell has the following inactive ingredients and dyes: gelatin, titanium dioxide, silicon dioxide and sodium lauryl sulfate. The chemical name for indinavir sulfate is [1(1S,2R),5(S)]-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[[(1,1 dimethylethyl)amino]carbonyl]-4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl)-D-erythro-pentonamide sulfate (1:1) salt. Indinavir sulfate has the following structural formula: Indinavir sulfate is a white to off-white, hygroscopic, crystalline powder with the molecular formula C36H47N5O4• H2SO4 and a molecular weight of 711.88. It is very soluble in water and in methanol. 1 Registered trademark of MERCK & CO., Inc. COPYRIGHT © 1996, 1997, 1998, 1999, 2004 MERCK & CO., Inc. All rights reserved MICROBIOLOGY Mechanism of Action HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Indinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. -

Prevalence of Drug Interactions in Hospitalized Elderly Patients: a Systematic Review
Supplementary material Eur J Hosp Pharm Prevalence of drug interactions in hospitalized elderly patients: a systematic review Luciana Mello de Oliveira 1,2; Juliana do Amaral Carneiro Diel1; Alessandra Nunes3; Tatiane da Silva Dal Pizzol 1,2,3 1Programa de Pós-Graduação em Epidemiologia, Faculdade de Medicina, Universidade Federal do Rio Grande do Sul. 2Programa de Pós-Graduação em Assistência Farmacêutica, Faculdade de Farmácia, Universidade Federal do Rio Grande do Sul. 3Faculdade de Farmácia, Universidade Federal do Rio Grande do Sul. Corresponding author: Luciana Mello de Oliveira – [email protected] and Tatiane da Silva Dal Pizzol - [email protected] Supplementary Table 3: Number of patients with interaction, number of DDI per patient with at least one DDI, drugs or drug classes mostly involved with DDI and drug combinations mostly involved with DDI. In cases which prevalence were described, we reported the three drugs mostly involved with drug interactions or the three drug combinations (or drug classes) mostly involved with DDI. ACE: angiotensin-converting enzyme. NA: not available. NSAID: non-steroidal anti-inflammatory drugs. PPI: proton-pump inhibitors. # of patients with # of DDI per patient with First autor interactions interaction Drugs or drug classes mostly involved with DDI Drug combinations mostly involved with DDI Barak-Tsarfir O, et al (61) Unclear: around 56 patients NA NA NA Warfarin; digitoxin; prednisolone antithrombotic agents; non-steroidal anti- 70 (evaluated only serious or inflammatory agents; angiotensin converting enzyme Blix HS, et al (29) contraindicated DDI) NA inhibitors N/A Serious: chlorpromazine + promethazine; chlorpromazine + haloperidol; haloperidol + promethazine; diazepam + phenobarbital; risperidone + haloperidol; carbamazepine + ketoconazole; carbamazepine + chlorpromazine; haloperidol + ketoconazole; chlorpromazine + ketoconazole; chlorpromazine + sodium phosphate. -

Ritonavir Mylan, INN-Ritonavir
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Ritonavir Mylan 100 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each film-coated tablet contains 100 mg of ritonavir. Excipient with known effect Each film-coated tablet contains 87.75 mg of sodium. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet (tablet). Yellow, capsule shaped, biconvex, beveled edge film-coated tablet, approximately 19.1 mm x 10.2 mm, debossed with ‘M163’ on one side and blank on the other side. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Ritonavir is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infected patients (adults and children of 2 years of age and older). 4.2 Posology and method of administration Ritonavir Mylan should be administered by physicians who are experienced in the treatment of HIV infection. Posology Ritonavir dosed as a pharmacokinetic enhancer When ritonavir is used as a pharmacokinetic enhancer with other protease inhibitors the Summary of Product Characteristics for the particular protease inhibitor must be consulted. The following HIV-1 protease inhibitors have been approved for use with ritonavir as a pharmacokinetic enhancer at the noted doses. Adults Amprenavir 600 mg twice daily with ritonavir 100 mg twice daily. Atazanavir 300 mg once daily with ritonavir 100 mg once daily. Fosamprenavir 700 mg twice daily with ritonavir 100 mg twice daily. Lopinavir co-formulated with ritonavir (lopinavir/ritonavir) 400 mg/100 mg or 800 mg/200 mg. Saquinavir 1,000 mg twice daily with ritonavir 100 mg twice daily in ART experienced patients. -

An Update on the Efficacy of Anti-Inflammatory Agents for Patients with Schizophrenia: Cambridge.Org/Psm a Meta-Analysis
Psychological Medicine An update on the efficacy of anti-inflammatory agents for patients with schizophrenia: cambridge.org/psm a meta-analysis 1,2 2,3,4 2,5 6 Review Article N. Çakici , N. J. M. van Beveren , G. Judge-Hundal , M. M. Koola and I. E. C. Sommer5 Cite this article: Çakici N, van Beveren NJM, Judge-Hundal G, Koola MM, Sommer IEC 1Department of Psychiatry and Amsterdam Neuroscience, Academic Medical Center, Meibergdreef 9, 1105 AZ (2019). An update on the efficacy of anti- Amsterdam, the Netherlands; 2Antes Center for Mental Health Care, Albrandswaardsedijk 74, 3172 AA, Poortugaal, inflammatory agents for patients with 3 schizophrenia: a meta-analysis. Psychological the Netherlands; Department of Psychiatry, Erasmus Medical Center, Doctor Molewaterplein 40, 3015 GD 4 Medicine 49, 2307–2319. https://doi.org/ Rotterdam, the Netherlands; Department of Neuroscience, Erasmus Medical Center, Doctor Molewaterplein 40, 5 10.1017/S0033291719001995 3015 GD Rotterdam, the Netherlands; Department of Psychiatry and Biomedical Sciences of Cells and Systems, University Medical Center Groningen, Deusinglaan 2, 9713AW Groningen, the Netherlands and 6Department of Received: 13 February 2019 Psychiatry and Behavioral Sciences, George Washington University School of Medicine and Health Sciences, 2300I Revised: 4 July 2019 St NW, Washington, DC 20052, USA Accepted: 16 July 2019 First published online: 23 August 2019 Abstract Key words: Background. Accumulating evidence shows that a propensity towards a pro-inflammatory Add-on antipsychotic therapy; estrogens; fatty acids; minocycline; N-acetylcysteine status in the brain plays an important role in schizophrenia. Anti-inflammatory drugs might compensate this propensity. This study provides an update regarding the efficacy of Author for correspondence: agents with some anti-inflammatory actions for schizophrenia symptoms tested in rando- N. -
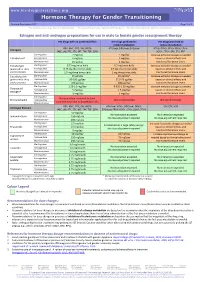
Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 for Personal Use Only
www.hiv-druginteractions.org Hormone Therapy for Gender Transitioning Revised September 2017 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Estrogen and anti-androgen preparations for use in male to female gender reassignment therapy HIV drugs with no predicted effect HIV drugs predicted to HIV drugs predicted to inhibit metabolism induce metabolism RPV, MVC, DTG, RAL, NRTIs ATV/cobi, DRV/cobi, EVG/cobi ATV/r, DRV/r, FPV/r, IDV/r, LPV/r, Estrogens (ABC, ddI, FTC, 3TC, d4T, TAF, TDF, ZDV) SQV/r, TPV/r, EFV, ETV, NVP Starting dose 2 mg/day 1 mg/day Increase estradiol dosage as needed Estradiol oral Average dose 4 mg/day 2 mg/day based on clinical effects and Maximum dose 8 mg/day 4 mg/day monitored hormone levels. Estradiol gel Starting dose 0.75 mg twice daily 0.5 mg twice daily Increase estradiol dosage as needed (preferred for >40 y Average dose 0.75 mg three times daily 0.5 mg three times daily based on clinical effects and and/or smokers) Maximum dose 1.5 mg three times daily 1 mg three times daily monitored hormone levels. Estradiol patch Starting dose 25 µg/day 25 µg/day* Increase estradiol dosage as needed (preferred for >40 y Average dose 50-100 µg/day 37.5-75 µg/day based on clinical effects and and/or smokers) Maximum dose 150 µg/day 100 µg/day monitored hormone levels. Starting dose 1.25-2.5 mg/day 0.625-1.25 mg/day Increase estradiol dosage as needed Conjugated Average dose 5 mg/day 2.5 mg/day based on clinical effects and estrogen† Maximum dose 10 mg/day 5 mg/day monitored hormone levels. -
Simvastatin 80Mg Tablets
Package leaflet: Information for the patient Simvastatin 80mg Tablets Read all of this leaflet carefully before you • if you are due to have an operation. You may need start taking this medicine because it contains to stop taking Simvastatin tablets for a short time important information for you. • if you are Asian, because a different dose may be • Keep this leaflet. You may need to read it again. applicable to you • If you have any further questions, ask your • if you are taking or have taken in the last 7 days doctor or pharmacist. a medicine called fusidic acid (a medicine for bacterial infection) orally or by injection. The • This medicine has been prescribed for you only. combination of fusidic acid and Simvastatin can Do not pass it on to others. It may harm them, lead to serious muscle problems (rhabdomyolysis). even if their signs of illness are the same as yours. Your doctor should do a blood test before you start • If you get any side effects, talk to your doctor taking Simvastatin and if you have any symptoms of or pharmacist. This includes any possible side liver problems while you take Simvastatin. This is to effects not listed in this leaflet. See section 4. check how well your liver is working. What is in this leaflet Your doctor may also want you to have blood tests to check how well your liver is working after you 1 What Simvastatin is and what it is used start taking Simvastatin. for 2 What you need to know before you take While you are on this medicine your doctor will Simvastatin monitor you closely if you have diabetes or are at risk of developing diabetes. -

Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis
Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis Centers for Disease Control and Prevention Office of Infectious Diseases National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination June 2013 This document is accessible online at http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Suggested citation: CDC. Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis [online]. 2013. Available from URL: http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Table of Contents Introduction 1 Methodology for Preparation of these Guidelines 2 The Role of Rifamycins in Tuberculosis Treatment 4 Managing Drug Interactions with Antivirals and Rifampin 5 Managing Drug Interactions with Antivirals and Rifabutin 9 Treatment of Latent TB Infection with Rifampin or Rifapentine 10 Treating Pregnant Women with Tuberculosis and HIV Co-infection 10 Treating Children with HIV-associated Tuberculosis 12 Co-treatment of Multidrug-resistant Tuberculosis and HIV 14 Limitations of these Guidelines 14 HIV-TB Drug Interaction Guideline Development Group 15 References 17 Table 1a. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in adults 21 Table 1b. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in children 22 Table 2a. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in adults 23 Table 2b. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in children 25 Table 3. Recommendations for co-administering antiretroviral drugs with RIFABUTIN in adults 26 ii Introduction Worldwide, tuberculosis is the most common serious opportunistic infection among people with HIV infection.