Polymer Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Title Crystallization of Stereospecific Olefin Copolymers (Special Issue on Physical Chemistry) Author(S) Sakaguchi, Fumio; Kita
Crystallization of Stereospecific Olefin Copolymers (Special Title Issue on Physical Chemistry) Author(s) Sakaguchi, Fumio; Kitamaru, Ryozo; Tsuji, Waichiro Bulletin of the Institute for Chemical Research, Kyoto Citation University (1966), 44(4): 295-315 Issue Date 1966-10-31 URL http://hdl.handle.net/2433/76134 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University Crystallization of Stereospecifie Olefin Copolymers Fumio SAKAGUCHI,Ryozo KITAMARU and Waichiro TSUJI* (Tsuji Laboratory) Received August 13, 1966 The stereoregularity of isotactic poly(4-methyl-1-pentene) was characterized and isomorphism phenomena were examined for the copolymeric systems of 4-methyl-1-pentene with several olefins in order to study the crystallization phenomena in these olefin copoly- mers polymerized with stereospecific catalysts. The structural heterogeneity or the fine crystalline structure of poly(4-methyl-1-pentene) could be correlated with its molecular structure by viewing this stereoregular homopolymer as if it were a copolymer. Cocrystallization or isomorphism phenomenon was recognized for the copolymeric systems of 4-methyl-1-pentene with butene-1, pentene-1, decene-1 and 3-methyl-1-butene, while no evidence of the phenomenon was obtained for the copolymeric systems with styrene and propylene. The degree of the isomorphism of those copolymers was discussed with the informations on the crystalline phases obtained from the X-ray study, on the constitution of the copolymeric chains in the amorphous phases obtained from the viscoelastic studies and on the other thermodynamical properties of these systems. INTRODUCTION Many works have been made with regard to the homopolymerization of olefins with stereospecific catalysts, i. e. complex catalysts composed of the combination of organometallic compound and transitional metallic compound. -

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

Properties and Synthesis. Elimination Reactions of Alkyl Halides
P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 7 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS. ELIMINATION REACTIONS OF ALKYL HALIDES SOLUTIONS TO PROBLEMS 7.1 (a) ( E )-1-Bromo-1-chloro-1-pentene or ( E )-1-Bromo-1-chloropent-1-ene (b) ( E )-2-Bromo-1-chloro-1-iodo-1-butene or ( E )-2-Bromo-1-chloro-1-iodobut-1-ene (c) ( Z )-3,5-Dimethyl-2-hexene or ( Z )-3,5-Dimethylhex-2-ene (d) ( Z )-1-Chloro-1-iodo-2-methyl-1-butene or ( Z )-1-Chloro-1-iodo-2-methylbut-1-ene (e) ( Z,4 S )-3,4-Dimethyl-2-hexene or ( Z,4 S )-3,4-Dimethylhex-2-ene (f) ( Z,3 S )-1-Bromo-2-chloro-3-methyl-1-hexene or (Z,3 S )-1-Bromo-2-chloro-3-methylhex-1-ene 7.2 < < Order of increasing stability 7.3 (a), (b) H − 2 ∆ H° = − 119 kJ mol 1 Pt 2-Methyl-1-butene pressure (disubstituted) H2 − ∆ H° = − 127 kJ mol 1 Pt 3-Methyl-1-butene pressure (monosubstituted) H2 − ∆ H° = − 113 kJ mol 1 Pt 2-Methyl-2-butene pressure (trisubstituted) (c) Yes, because hydrogenation converts each alkene into the same product. 106 CONFIRMING PAGES P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS 107 H H (d) > > H H H (trisubstituted) (disubstituted) (monosubstituted) Notice that this predicted order of stability is confirmed by the heats of hydro- genation. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -
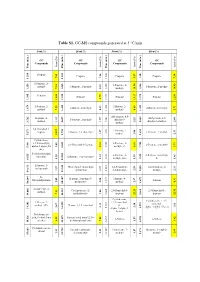
Table S1. GC-MS Compounds Generated at 5 °C/Min
Table S1. GC-MS compounds generated at 5 °C/min. 0 wt.% 10 wt.% 30 wt.% 50 wt.% GC GC GC GC Compounds Compounds Compounds Compounds Area (%) Area (%) Area (%) Area (%) Time (min.) (min.) Time Time (min.) (min.) Time (min.) Time (min.) Time Propene Propene Propene Propene 7.42 7.42 1.26 5.47 1.20 7.38 1.200 1.200 1.200 1.214 1.214 1-Propene, 2- 1-Propene, 2- methyl- 1-Propene, 2-methyl- 1-Propene, 2-methyl- 7.34 7.34 2.41 methyl- 1.245 1.245 1.245 1.259 1.259 10.54 1.246 14.90 Pentane Pentane Pentane Pentane 3.79 3.79 1.375 1.375 1.395 1.395 11.81 1.381 13.17 1.375 12.83 1-Pentene, 2- 2-Butene, 2- 2-Butene, 2-methyl- 2-Butene, 2-methyl- methyl- 7.25 0.93 methyl- 4.65 5.71 1.426 1.426 1.420 1.420 1.679 1.679 1H-Pyrazole, 4,5- Heptane, 4- 1H-Pyrazole, 4,5- 1-Pentene, 2-methyl- dihydro-5- methyl- 1.92 1.57 2.25 dihydro-5-methyl- 2.44 1.659 1.659 1.562 1.569 3.762 3.762 methyl- 2,4-Dimethyl-1- 1-Pentene, 2- heptene 2-Butene, 2,3-dimethyl- 1-Pentene, 2-methyl- 1.27 1.27 methyl- 3.08 2.33 1.653 1.653 1.653 5.671 5.671 34.57 1.750 Cyclohexane, 1,3,5-trimethyl-, 2-Pentene, 3- 2,4-Dimethyl-1-heptene 2-Pentene, 2-methyl- alpha.,3.alpha.,5.b 2.03 9.11 methyl-, €- 6.11 5.73 5.605 5.605 1.743 1.744 5.981 5.981 eta.)- 5-Aminoisoxazole 2-Pentene, 3- 2-Pentene, 3-methyl-, Spermine Ethanone, 1-cyclopentyl- 1.55 1.55 1.32 methyl-, (Z)- 1.59 €- 1.46 1.828 1.828 1.828 6.925 6.925 10.560 Ethanone, 1- 1R,2c,3t,4t-Tetramethyl- 1,3-Pentadiene, 1,4-Hexadiene, 5- cyclopentyl- 3.57 3.57 cyclohexan 0.97 2,3-dimethyl- 2.28 methyl- 2.31 2.584 2.584 2.585 10.613 10.613 10.638 N- Heptane, 2-methyl-3- Heptane, 4- Methylallylamine Toluene 2.49 2.49 methylene- 2.50 methyl- 2.97 4.31 3.697 3.697 3.697 14.047 14.047 10.684 10.684 2-Undecene, 4- Cyclopentane, (2- 2,4-Dimethyl-1- 2,4-Dimethyl-1- methyl- 7.81 7.81 methylbutyl)- 1.00 heptene heptene 5.605 5.605 22.29 22.29 5.605 12.95 14.086 14.086 14.163 Cyclohexane, Cyclohexane, 1,3,5- 2-Decene, 7- 1,3,5-trimethyl-, trimethyl- methyl-, (Z)- Hexane, 2,3,4-trimethyl- (1. -

Gfsorganics & Fragrances
Chemicals for Flavors GFSOrganics & Fragrances GFS offers a broad range of specialty organic chemicals Specialized chemistries we as building blocks and intermediates for the manufacture of offer include: flavors and fragrances. • Alkynes Over 5,500 materials, including 1,400 chemicals from natural sources, are used for flavor • Alkynols enhancements and aroma profiles. These aroma chemicals are integral components of • Olefins the continued growth within consumer products and packaged foods. The diversity of products can be attributed to the broad spectrum of organic compounds derived from • Unsaturated Acids esters, aldehydes, lactones, alcohols and several other functional groups. • Unsaturated Esters • DIPPN and other Products GFS Chemicals manufactures a wide range of organic intermediates that have been utilized in a multitude of personal care applications. For example, we support Why GFS? several market leading companies in the manufacture and supply of alkyne based aroma chemicals. • Specialized Chemistries • Tailored Specifications As such, we understand the demanding nature of this fast changing market and are fully • From Grams to Metric Tons equipped to overcome process challenges and manufacture the novel chemical products • Responsive Technical Staff that you need, when you need them. We offer flexible custom manufacturing services to produce high purity products with the assurance of quality and full confidentiality. • Uncompromised Product Quality Our state-of-the-art manufacturing facility, located in Columbus, OH is ISO 9001:2008 certified. As a U.S. based manufacturer we have a proven record of helping you take products from development to commercialization. Our technical sales experts are readily accessible to discuss your project needs and unique product specifications. -

Ionic Liquid*
Pure Appl. Chem., Vol. 83, No. 7, pp. 1391–1406, 2011. doi:10.1351/PAC-CON-10-12-03 © 2011 IUPAC, Publication date (Web): 10 June 2011 Oligomerisation of linear 1-olefins using a chlorogallate(III) ionic liquid* Martin P. Atkins1, Kenneth R. Seddon2,‡, and Małgorzata Swadźba-Kwaśny2,‡ 1PETRONAS Research Sdn Bhd, Lot 3288&3289, Off Jalan Ayer Itam, Kawasan Institusi Bangi, 43000, Kajang, Malaysia; 2QUILL, The Queen’s University of Belfast, Belfast, BT9 5AG, UK Abstract: A Lewis acidic chlorogallate(III) ionic liquid, 1-ethyl-3-methylimidazolium hepta - chlorodigallate(III), [C2mim][Ga2Cl7], was successfully used to oligomerise 1-pentene. The influence of temperature, time, catalyst concentration, and stirring rate on conversion and product distribution was modelled using a design of experiment (DoE) approach (chemo - metrics). The process was optimised for lubricant base oils production; the C20–C50 fraction (where Cn indicates the number of carbons in the oligomer) was maximised, while the heav- ier oligomer fraction (>C50) was minimised. Keywords: 1-alkenes; chemometrics; ionic liquids; linear terminal olefins; 1-olefins; oligomerisation. INTRODUCTION Oligomerisation and ionic liquids Polyalphaolefins (PAOs), used as a base for synthetic automotive lubricants (Group 4 base stock by API classification) [1], are traditionally made by oligomerisation of 1-decene to C20–C50 blend (where Cn indicates the number of carbons in the oligomer) [2] using Ziegler–Natta or Friedel–Crafts catalysts [3]. The carbon number is important, as the pour point of higher polymers (>C50) is too high, whilst the lower cut (<C20) is too volatile. 1-Decene is manufactured in a very selective manner from ethene [2]. -

Steam Cracking: Chemical Engineering
Steam Cracking: Kinetics and Feed Characterisation João Pedro Vilhena de Freitas Moreira Thesis to obtain the Master of Science Degree in Chemical Engineering Supervisors: Professor Doctor Henrique Aníbal Santos de Matos Doctor Štepánˇ Špatenka Examination Committee Chairperson: Professor Doctor Carlos Manuel Faria de Barros Henriques Supervisor: Professor Doctor Henrique Aníbal Santos de Matos Member of the Committee: Specialist Engineer André Alexandre Bravo Ferreira Vilelas November 2015 ii The roots of education are bitter, but the fruit is sweet. – Aristotle All I am I owe to my mother. – George Washington iii iv Acknowledgments To begin with, my deepest thanks to Professor Carla Pinheiro, Professor Henrique Matos and Pro- fessor Costas Pantelides for allowing me to take this internship at Process Systems Enterprise Ltd., London, a seven-month truly worthy experience for both my professional and personal life which I will certainly never forget. I would also like to thank my PSE and IST supervisors, who help me to go through this final journey as a Chemical Engineering student. To Stˇ epˇ an´ and Sreekumar from PSE, thank you so much for your patience, for helping and encouraging me to always keep a positive attitude, even when harder problems arose. To Prof. Henrique who always showed availability to answer my questions and to meet in person whenever possible. Gostaria tambem´ de agradecer aos meus colegas de casa e de curso Andre,´ Frederico, Joana e Miguel, com quem partilhei casa. Foi uma experienciaˆ inesquec´ıvel que atravessamos´ juntos e cer- tamente que a vossa presenc¸a diaria´ apos´ cada dia de trabalho ajudou imenso a aliviar as saudades de casa. -

Olefins from Biomass Intermediates: a Review
catalysts Review Review OlefinsOlefins fromfrom BiomassBiomass Intermediates:Intermediates: AA ReviewReview 1 1,2, VasilikiVasiliki Zacharopoulou Zacharopoulou1 andand AngelikiAngeliki A.A. LemonidouLemonidou 1,2,** 1 Department of Chemical Engineering, Aristotle University of Thessaloniki, University Campus, 1 Department of Chemical Engineering, Aristotle University of Thessaloniki, University Campus, Thessaloniki 54124, Greece; [email protected] 54124 Thessaloniki, Greece; [email protected] 2 Chemical Process Engineering Research Institute (CERTH/CPERI), P.O. Box 60361 Thermi, 2 Chemical Process Engineering Research Institute (CERTH/CPERI), P.O. Box 60361 Thermi, Thessaloniki 57001, Greece 57001 Thessaloniki, Greece * Correspondence: [email protected]; Tel.: +30‐2310‐996‐273 * Correspondence: [email protected]; Tel.: +30-2310-996-273 Received: 16 November 2017; Accepted: 19 December 2017; Published: Received: 16 November 2017; Accepted: 19 December 2017; Published: 23 December 2017 Abstract:Abstract: Over the last decade, increasing demand for olefinsolefins and theirtheir valuablevaluable productsproducts hashas promptedprompted researchresearch onon novelnovel processesprocesses andand technologiestechnologies forfor theirtheir selectiveselective production.production. As olefinsolefins are predominatelypredominately dependent on fossil resources, their production is limited by thethe finitefinite reservesreserves and the associated economic and environmental concerns.concerns. The need for alternative routes for -

Nuclear Spin-Induced Optical Rotation of Functional Groups in Hydrocarbons Petr Štěpánek1 Supplementary Information
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is © the Owner Societies 2020 Nuclear spin-induced optical rotation of functional groups in hydrocarbons Petr Štěpánek1 Supplementary Information Contents 1 List of molecules 2 2 Basis set benchmark 5 1 3 Effect of cis/trans isomerism on H NSOR of T1 atom type in alkenes 5 4 Effect of conformation on NSOR 6 5 Extended Figure for NSOR of dienes 7 6 Effect of cis/trans isomerism in dienes 8 7 Supporting data for solvent and geometry effects 8 1NMR Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, FI-90014 Oulu, Finland; petr.stepanek@oulu.fi 1 1 List of molecules Tables below list the molecules included in the study. The test cases of the contribution model are not included. Table S 1: Alkane molecules methane 2-methyl-propane 2,3-dimethyl-butane ethane 2-methyl-butane 2,2-dimethyl-propane propane 2-methyl-pentane 2,2-dimethyl-pentane butane 3-methyl-pentane 2,3-dimethyl-pentane pentane 2,2-dimethyl-butane 3,3-dimethyl-pentane hexane Table S 2: Alkene molecules ethene 2,3,3-trimethyl-butene 5-methyl-heptene propene 2,4-dimethyl-pentene 6-methyl-heptene trans-but-2-ene 2-methyl-hexene cis-5,5-dimethyl-hex-2-ene cis-pent-2-ene 3,3-dimethyl-pentene cis-6-methyl-hept-2-ene trans-hex-2-ene 3,4-dimethyl-pentene trans-5,5-dimethyl-hex-2-ene trans-pent-2-ene 3-methyl-hexene trans-6-methyl-hept-2-ene trans-hex-3-ene 4-methyl-hexene 5,6-dimethyl-heptene 2-methyl-propene cis-4,4-dimethyl-pent-2-ene cis-6,6-dimethyl-hepte-2-ene 2-methyl-butene -

Carbene Rearrangements: Intramolecular Interaction of a Triple Bond with a Carbene Center
An Abstract OF THE THESIS OF Jose C. Danino for the degree of Doctor of Philosophy in Chemistry presented on _Dcc, Title: Carbene RearrangementE) Intramolecular Interaction of a Triple Bond with aCarbene Center Redacted for Privacy Abstract approved: Dr. Vetere. Freeman The tosylhydrazones of2-heptanone, 4,4-dimethy1-2- heptanone, 6-heptyn-2-one and 4,4-dimethy1-6-heptyn-2- one were synthesizedand decomposed under a varietyof reaction conditions:' drylithium and sodium salt pyrolyses, sodium methoxide thermolysesin diglyme and photolyses of the lithium salt intetrahydrofuran. The saturated ana- logues 2-heptanone tosylhydrazoneand its 4,4-dimethyl isomer afforded the alkenesarising from 6-hydrogeninser- product distribution in the tion. It was determined that differ- dry salt pyrolyses of2-heptanone tosylhydrazone was ent for the lithiumand the sodium salts. However, the product distribution of thedry sodium salt was verysimilar diglyme to product distributionobtained on thermolysis in explained by a with sodium methoxide. This difference was reaction of lithium bromide(present as an impurity inall compound to the lithium salts)with the intermediate diazo afford an organolithiumintermediate that behaves in a some- what different fashionthan the free carbene.The unsaturated analogues were found to produce a cyclic product in addition to the expected acyclic alkenes arising from 3-hydrogen insertion. By comparison of the acyclic alkene distri- bution obtained in the saturated analogues with those in the unsaturated analogues, it was concluded that at leastsome cyclization was occurring via addition of the diazo moiety to the triple bond. It was determined that the organo- lithium intermediateresulting from lithium bromide cat- alyzed decomposition of the diazo compound was incapable of cyclization.