Rearrangements of Cyclopropyl and Related Carbenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Title Crystallization of Stereospecific Olefin Copolymers (Special Issue on Physical Chemistry) Author(S) Sakaguchi, Fumio; Kita
Crystallization of Stereospecific Olefin Copolymers (Special Title Issue on Physical Chemistry) Author(s) Sakaguchi, Fumio; Kitamaru, Ryozo; Tsuji, Waichiro Bulletin of the Institute for Chemical Research, Kyoto Citation University (1966), 44(4): 295-315 Issue Date 1966-10-31 URL http://hdl.handle.net/2433/76134 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University Crystallization of Stereospecifie Olefin Copolymers Fumio SAKAGUCHI,Ryozo KITAMARU and Waichiro TSUJI* (Tsuji Laboratory) Received August 13, 1966 The stereoregularity of isotactic poly(4-methyl-1-pentene) was characterized and isomorphism phenomena were examined for the copolymeric systems of 4-methyl-1-pentene with several olefins in order to study the crystallization phenomena in these olefin copoly- mers polymerized with stereospecific catalysts. The structural heterogeneity or the fine crystalline structure of poly(4-methyl-1-pentene) could be correlated with its molecular structure by viewing this stereoregular homopolymer as if it were a copolymer. Cocrystallization or isomorphism phenomenon was recognized for the copolymeric systems of 4-methyl-1-pentene with butene-1, pentene-1, decene-1 and 3-methyl-1-butene, while no evidence of the phenomenon was obtained for the copolymeric systems with styrene and propylene. The degree of the isomorphism of those copolymers was discussed with the informations on the crystalline phases obtained from the X-ray study, on the constitution of the copolymeric chains in the amorphous phases obtained from the viscoelastic studies and on the other thermodynamical properties of these systems. INTRODUCTION Many works have been made with regard to the homopolymerization of olefins with stereospecific catalysts, i. e. complex catalysts composed of the combination of organometallic compound and transitional metallic compound. -

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -

Properties and Synthesis. Elimination Reactions of Alkyl Halides
P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 7 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS. ELIMINATION REACTIONS OF ALKYL HALIDES SOLUTIONS TO PROBLEMS 7.1 (a) ( E )-1-Bromo-1-chloro-1-pentene or ( E )-1-Bromo-1-chloropent-1-ene (b) ( E )-2-Bromo-1-chloro-1-iodo-1-butene or ( E )-2-Bromo-1-chloro-1-iodobut-1-ene (c) ( Z )-3,5-Dimethyl-2-hexene or ( Z )-3,5-Dimethylhex-2-ene (d) ( Z )-1-Chloro-1-iodo-2-methyl-1-butene or ( Z )-1-Chloro-1-iodo-2-methylbut-1-ene (e) ( Z,4 S )-3,4-Dimethyl-2-hexene or ( Z,4 S )-3,4-Dimethylhex-2-ene (f) ( Z,3 S )-1-Bromo-2-chloro-3-methyl-1-hexene or (Z,3 S )-1-Bromo-2-chloro-3-methylhex-1-ene 7.2 < < Order of increasing stability 7.3 (a), (b) H − 2 ∆ H° = − 119 kJ mol 1 Pt 2-Methyl-1-butene pressure (disubstituted) H2 − ∆ H° = − 127 kJ mol 1 Pt 3-Methyl-1-butene pressure (monosubstituted) H2 − ∆ H° = − 113 kJ mol 1 Pt 2-Methyl-2-butene pressure (trisubstituted) (c) Yes, because hydrogenation converts each alkene into the same product. 106 CONFIRMING PAGES P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS 107 H H (d) > > H H H (trisubstituted) (disubstituted) (monosubstituted) Notice that this predicted order of stability is confirmed by the heats of hydro- genation. -

Revised Group Additivity Values for Enthalpies of Formation (At 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds
Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds Cite as: Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 Submitted: 17 January 1996 . Published Online: 15 October 2009 N. Cohen ARTICLES YOU MAY BE INTERESTED IN Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties The Journal of Chemical Physics 29, 546 (1958); https://doi.org/10.1063/1.1744539 Critical Evaluation of Thermochemical Properties of C1–C4 Species: Updated Group- Contributions to Estimate Thermochemical Properties Journal of Physical and Chemical Reference Data 44, 013101 (2015); https:// doi.org/10.1063/1.4902535 Estimation of the Thermodynamic Properties of Hydrocarbons at 298.15 K Journal of Physical and Chemical Reference Data 17, 1637 (1988); https:// doi.org/10.1063/1.555814 Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 25, 1411 © 1996 American Institute of Physics for the National Institute of Standards and Technology. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds N. Cohen Thermochemical Kinetics Research, 6507 SE 31st Avenue, Portland, Oregon 97202-8627 Received January 17, 1996; revised manuscript received September 4, 1996 A program has been undertaken for the evaluation and revision of group additivity values (GAVs) necessary for predicting, by means of Benson's group additivity method, thermochemical properties of organic molecules. This review reports on the portion of that program dealing with GAVs for enthalpies of formation at 298.15 K (hereinafter abbreviated as 298 K) for carbon-hydrogen and carbon-hydrogen-oxygen compounds. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -
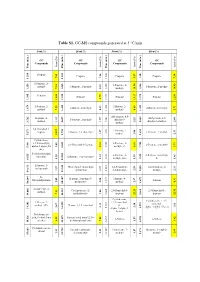
Table S1. GC-MS Compounds Generated at 5 °C/Min
Table S1. GC-MS compounds generated at 5 °C/min. 0 wt.% 10 wt.% 30 wt.% 50 wt.% GC GC GC GC Compounds Compounds Compounds Compounds Area (%) Area (%) Area (%) Area (%) Time (min.) (min.) Time Time (min.) (min.) Time (min.) Time (min.) Time Propene Propene Propene Propene 7.42 7.42 1.26 5.47 1.20 7.38 1.200 1.200 1.200 1.214 1.214 1-Propene, 2- 1-Propene, 2- methyl- 1-Propene, 2-methyl- 1-Propene, 2-methyl- 7.34 7.34 2.41 methyl- 1.245 1.245 1.245 1.259 1.259 10.54 1.246 14.90 Pentane Pentane Pentane Pentane 3.79 3.79 1.375 1.375 1.395 1.395 11.81 1.381 13.17 1.375 12.83 1-Pentene, 2- 2-Butene, 2- 2-Butene, 2-methyl- 2-Butene, 2-methyl- methyl- 7.25 0.93 methyl- 4.65 5.71 1.426 1.426 1.420 1.420 1.679 1.679 1H-Pyrazole, 4,5- Heptane, 4- 1H-Pyrazole, 4,5- 1-Pentene, 2-methyl- dihydro-5- methyl- 1.92 1.57 2.25 dihydro-5-methyl- 2.44 1.659 1.659 1.562 1.569 3.762 3.762 methyl- 2,4-Dimethyl-1- 1-Pentene, 2- heptene 2-Butene, 2,3-dimethyl- 1-Pentene, 2-methyl- 1.27 1.27 methyl- 3.08 2.33 1.653 1.653 1.653 5.671 5.671 34.57 1.750 Cyclohexane, 1,3,5-trimethyl-, 2-Pentene, 3- 2,4-Dimethyl-1-heptene 2-Pentene, 2-methyl- alpha.,3.alpha.,5.b 2.03 9.11 methyl-, €- 6.11 5.73 5.605 5.605 1.743 1.744 5.981 5.981 eta.)- 5-Aminoisoxazole 2-Pentene, 3- 2-Pentene, 3-methyl-, Spermine Ethanone, 1-cyclopentyl- 1.55 1.55 1.32 methyl-, (Z)- 1.59 €- 1.46 1.828 1.828 1.828 6.925 6.925 10.560 Ethanone, 1- 1R,2c,3t,4t-Tetramethyl- 1,3-Pentadiene, 1,4-Hexadiene, 5- cyclopentyl- 3.57 3.57 cyclohexan 0.97 2,3-dimethyl- 2.28 methyl- 2.31 2.584 2.584 2.585 10.613 10.613 10.638 N- Heptane, 2-methyl-3- Heptane, 4- Methylallylamine Toluene 2.49 2.49 methylene- 2.50 methyl- 2.97 4.31 3.697 3.697 3.697 14.047 14.047 10.684 10.684 2-Undecene, 4- Cyclopentane, (2- 2,4-Dimethyl-1- 2,4-Dimethyl-1- methyl- 7.81 7.81 methylbutyl)- 1.00 heptene heptene 5.605 5.605 22.29 22.29 5.605 12.95 14.086 14.086 14.163 Cyclohexane, Cyclohexane, 1,3,5- 2-Decene, 7- 1,3,5-trimethyl-, trimethyl- methyl-, (Z)- Hexane, 2,3,4-trimethyl- (1. -

Structure and Bonding of New Boron and Carbon Superpolyhedra
Structural Chemistry (2019) 30:805–814 https://doi.org/10.1007/s11224-019-1279-5 ORIGINAL RESEARCH Structure and bonding of new boron and carbon superpolyhedra Olga A. Gapurenko1 & Ruslan M. Minyaev1 & Nikita S. Fedik2 & Vitaliy V. Koval1 & Alexander I. Boldyrev2 & Vladimir I. Minkin1 Received: 16 November 2018 /Accepted: 1 January 2019 /Published online: 10 January 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Using the DFT methods, we computationally predict the stability of cage compounds E4nRn (E = B, C; R = H, F; n = 4, 8, 12, 24) based on Platonic bodies and Archimedean polyhedrons in which all vertices are replaced by tetrahedral E4R fragments. Cage compounds B60R12 and C60 with pyramidal units B5RorC5 are also examined and it is shown that only boron compounds are stable. The nature of chemical bonding in the discussed compounds is analyzed using the AdNDP and NBO methods. The hydrocarbons have classical 2c-2e C-C σ-bonds, while the boron compounds are formed by the polyhedral units with the delocalized multicenter bonds which connected three and more boron atoms. The new example of spherical aromaticity accord- 2 ing to the 2(N+1) rule in the case of B16F4 with multicenter 16c-2e bonds are revealed. Stable compound B60H12 contains 12 5c- 2e B-B bonds. Keywords Сage clusters . Chemical bonding . 3c-2e bond . Spherical aromaticity . AdNDP . NBO Construction of novel allotropic forms of carbon based on was proposed [1] as the system with the same symmetry as the tetrahedrane- and cubane-like building blocks was pro- sp3-carbon to replace the carbon atoms in the diamond posed by Burdett and Lee [1] and by Johnston and lattice. -

Gfsorganics & Fragrances
Chemicals for Flavors GFSOrganics & Fragrances GFS offers a broad range of specialty organic chemicals Specialized chemistries we as building blocks and intermediates for the manufacture of offer include: flavors and fragrances. • Alkynes Over 5,500 materials, including 1,400 chemicals from natural sources, are used for flavor • Alkynols enhancements and aroma profiles. These aroma chemicals are integral components of • Olefins the continued growth within consumer products and packaged foods. The diversity of products can be attributed to the broad spectrum of organic compounds derived from • Unsaturated Acids esters, aldehydes, lactones, alcohols and several other functional groups. • Unsaturated Esters • DIPPN and other Products GFS Chemicals manufactures a wide range of organic intermediates that have been utilized in a multitude of personal care applications. For example, we support Why GFS? several market leading companies in the manufacture and supply of alkyne based aroma chemicals. • Specialized Chemistries • Tailored Specifications As such, we understand the demanding nature of this fast changing market and are fully • From Grams to Metric Tons equipped to overcome process challenges and manufacture the novel chemical products • Responsive Technical Staff that you need, when you need them. We offer flexible custom manufacturing services to produce high purity products with the assurance of quality and full confidentiality. • Uncompromised Product Quality Our state-of-the-art manufacturing facility, located in Columbus, OH is ISO 9001:2008 certified. As a U.S. based manufacturer we have a proven record of helping you take products from development to commercialization. Our technical sales experts are readily accessible to discuss your project needs and unique product specifications. -

Ring-Opening Alkyne Metathesis Methods for Functional Conjugated Polymer Synthesis by Stephen W. Von Kugelgen a Dissertation
Ring-Opening Alkyne Metathesis Methods For Functional Conjugated Polymer Synthesis by Stephen W. von Kugelgen A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Felix R. Fischer, Chair Professor Robert G. Bergman Professor Phillip B. Messersmith Fall 2018 Ring-Opening Alkyne Metathesis Methods For Functional Conjugated Polymer Synthesis Copyright 2018 by Stephen W. von Kugelgen 1 Abstract Ring-Opening Alkyne Metathesis Methods For Functional Conjugated Polymer Synthesis by Stephen W. von Kugelgen Doctor of Philosophy in Chemistry University of California, Berkeley Professor Felix R. Fischer, Chair Since its discovery in the mid 20th century, most applications of alkyne metathesis have relied on thermodynamics to control product distributions. Ring-opening alkyne metathe- sis polymerization (ROAMP), in contrast, requires the kinetic product of metathesis of a strained, cyclic alkyne monomer to give a living, chain-growth polymerization (Chapter 1, Introduction). This living polymerization of conjugated alkyne-containing monomers has the potential to access a wide range of functional poly(arylene ethynylene) materials with excep- tional control over length, dispersity, topology, and endgroups. To this end, we demonstrate the first ROAMP synthesis of conjugated poly(ortho-phenylene ethynylene) and elucidate a mechanistic description of the reaction to understand the enabling catalyst selectivity and unexpectely find that initiator sterics dictate endgroup fidelity and polymer topology (Chap- ter 2). To disentangle the role of steric and electronic factors in initiator performance, we describe a novel synthetic method that gives a series of isosteric benzylidyne catalysts which exhibit a strong, deterministic electronic effect on both ROAMP initiation rates and polymer endgroup fidelity (Chapter 3). -

Synthesis and Reactivity of Small Ring Bicyclic Compounds Raymond Lee Welch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1969 Synthesis and reactivity of small ring bicyclic compounds Raymond Lee Welch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Welch, Raymond Lee, "Synthesis and reactivity of small ring bicyclic compounds " (1969). Retrospective Theses and Dissertations. 3807. https://lib.dr.iastate.edu/rtd/3807 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 70-7767 WELCH, Raymond Lee, 1948- SYNTHESIS AND REACTIVITY OF SMALL RING BICYCLIC COMPOUNDS. Iowa State University, PhJD., 1969 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESIS AND REACTIVITY OF SMALL RING BICYCLIC COMPOUNDS by Raymond Lee Welch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. In Charge of Major Work Signature was redacted for privacy. Signature was redacted for privacy. Iowa State University Of Science and Technology