UCLA Previously Published Works
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Title of the Article
Mechanism-Anchored Profiling Derived from Epigenetic Networks Predicts Outcome in Acute Lymphoblastic Leukemia Xinan Yang, PhD1, Yong Huang, MD1, James L Chen, MD1, Jianming Xie, MSc2, Xiao Sun, PhD2, Yves A Lussier, MD1,3,4§ 1Center for Biomedical Informatics and Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, IL 60637 USA 2State Key Laboratory of Bioelectronics, Southeast University, 210096 Nanjing, P.R.China 3The University of Chicago Cancer Research Center, and The Ludwig Center for Metastasis Research, The University of Chicago, Chicago, IL 60637 USA 4The Institute for Genomics and Systems Biology, and the Computational Institute, The University of Chicago, Chicago, IL 60637 USA §Corresponding author Email addresses: XY: [email protected] YH: [email protected] JC: [email protected] JX: [email protected] XS: [email protected] YL: [email protected] - 1 - Abstract Background Current outcome predictors based on “molecular profiling” rely on gene lists selected without consideration for their molecular mechanisms. This study was designed to demonstrate that we could learn about genes related to a specific mechanism and further use this knowledge to predict outcome in patients – a paradigm shift towards accurate “mechanism-anchored profiling”. We propose a novel algorithm, PGnet, which predicts a tripartite mechanism-anchored network associated to epigenetic regulation consisting of phenotypes, genes and mechanisms. Genes termed as GEMs in this network meet all of the following criteria: (i) they are co-expressed with genes known to be involved in the biological mechanism of interest, (ii) they are also differentially expressed between distinct phenotypes relevant to the study, and (iii) as a biomodule, genes correlate with both the mechanism and the phenotype. -

Identification and Diagnostic Performance of a Small RNA Within the PCA3 and BMCC1 Gene Locus That Potentially Targets Mrna
Published OnlineFirst November 12, 2014; DOI: 10.1158/1055-9965.EPI-14-0377 Research Article Cancer Epidemiology, Biomarkers Identification and Diagnostic Performance of a & Prevention Small RNA within the PCA3 and BMCC1 Gene Locus That Potentially Targets mRNA Ross M. Drayton1, Ishtiaq Rehman1, Raymond Clarke2, Zhongming Zhao3,4, Karl Pang1, Saiful Miah1, Robert Stoehr5, Arndt Hartmann5, Sheila Blizard1, Martin Lavin2, Helen E. Bryant1, Elena S. Martens-Uzunova6, Guido Jenster6, Freddie C. Hamdy7, Robert A. Gardiner2, and James W.F. Catto1 Abstract Background: PCA3 is a long noncoding RNA (lncRNA) with malignant prostatic tissues, exfoliated urinary cells from men unknown function, upregulated in prostate cancer. LncRNAs may with prostate cancer (13–273 fold change; t test P < 0.003), and be processed into smaller active species. We hypothesized this for closely correlated to PCA3 expression (r ¼ 0.84–0.93; P < 0.001). PCA3. Urinary PCA3-shRNA2 (C-index, 0.75–0.81) and PCA3 (C-index, Methods: We computed feasible RNA hairpins within the 0.78) could predict the presence of cancer in most men. PCA3- BMCC1 gene (encompassing PCA3) and searched a prostate shRNA2 knockup altered the expression of predicted target transcriptome for these. We measured expression using qRT- mRNAs, including COPS2, SOX11, WDR48, TEAD1, and Noggin. PCR in three cohorts of prostate cancer tissues (n ¼ 60), PCA3-shRNA2 expression was negatively correlated with COPS2 exfoliated urinary cells (n ¼ 484 with cancer and n ¼ 166 in patient samples (r ¼0.32; P < 0.001). controls), and in cell lines (n ¼ 22). We used in silico predictions Conclusion: We identified a short RNA within PCA3, whose and RNA knockup to identify potential mRNA targets of short expression is correlated to PCA3, which may target mRNAs transcribed RNAs. -

Supplementary Material for “Characterization of the Opossum Immune Genome Provides Insights Into the Evolution of the Mammalian Immune System”
Supplementary material for “Characterization of the opossum immune genome provides insights into the evolution of the mammalian immune system” Katherine Belov1*, Claire E. Sanderson1, Janine E. Deakin2, Emily S.W. Wong1, Daniel Assange3, Kaighin A. McColl3, Alex Gout3,4, Bernard de Bono5, Terence P. Speed3, John Trowsdale5, Anthony T. Papenfuss3 1. Faculty of Veterinary Science, University of Sydney, Sydney, Australia 2. ARC Centre for Kangaroo Genomics, Research School of Biological Sciences, The Australian National University, Canberra, Australia 3. Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Australia 4. Department of Medical Biology, The University of Melbourne, Parkville, Australia 5. Immunology Division, University of Cambridge, Cambridge, UK *Corresponding author: K. Belov, Faculty of Veterinary Science, University of Sydney, NSW 2006, Australia ph 61 2 9351 3454, fx 61 2 9351 3957, email [email protected] MHC paralogous regions Only 36 of the 114 genes in the opossum MHC have paralogs in one of the three paralogous regions (Supplementary Table 1). Genes represented in at least three of the four paralogous regions (13 genes) were used to compare gene order, revealing rearrangements between the four regions in opossum. Table 1: MHC genes with paralogs on opossum chromosomes 1, 2 and 3, corresponding to MHC paralogous regions on human chromosomes 9, 1 and 19 respectively. MHC Chromosome 1 Chromosome 2 Chromosome 3 (Human Chr 9) (Human Chr 1) (Human Chr 19) AGPAT1 AGPAT2 AIF1 C9orf58 ATP6V1G2 ATP6V1G1 ATP6V1G3 B3GALT4 B3GALT2 BAT1 DDX39 BAT2 KIAA0515 BAT2D1 BRD2 BRD3 BRDT BRD4 C4 C5 C3 SLC44A4 SLC44A5 SLC44A2 CLIC1 CLIC3 CLIC4 COL11A2 COL5A1 COL11A1 COL5A3 CREBL1 ATF6 DDAH2 DDAH1 DDR1 DDR2 EGFL8 EGFL7 EHMT2 EHMT1 GPX5 GPX4 MHC Class I CD1 HSPA1A HSPA5 MDC1 PRG4 NOTCH4 NOTCH1 NOTCH2 NOTCH3 PBX2 PBX3 PBX1 PBX4 PHF1 MTF2 PRSS16 DPP7 PSMB9 PSMB7 RGL2 RALGDS RGL1 RGL3 RING1 RNF2 RXRB RXRA RXRG SYNGAP1 RASAL2 TAP ABCA2 TNF/LTA/LTB TNFSF8/TNFSF15 TNFSF4 CD70/TNFSF9/ TNFSF14/ TNXB TNC TNR Table 2. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Examination of the Role of the Hu Proteins, Hur and Hud, in Prostate Cancer Cells
Examination of the Role of the Hu Proteins, HuR and HuD, in Prostate Cancer Cells. Christin Florence Down This thesis is presented for the degree of Doctor of Philosophy to the School of Medicine and Pharmacology, University of Western Australia. January 2007 i Declaration. This thesis contains published work and/or work prepared for publication, some of which has been co-authored . The bibliographic details of the works and where they appear in the thesis are set out below. (The candidate must attach to this declaration a statement detailing the percentage contribution of each author to the work. This must been signed by all authors. Where this is not possible, the statement detailing the percentage contribution of authors should be signed by the candidate’s Coordinating Supervisor). This thesis contains experimental data from the following co-authored work: Hu Proteins are Expressed and Regulate Androgen Receptor Expression and Cell Proliferation in Prostate Cancer. Christin F Down , Dianne J Beveridge, Michael R Epis, Ricky Lareu, Lisa M Stuart, Britt Granath, Dominic C Voon, Henry Furneaux, Cecily Metcalf, Jacqueline Bentel and Peter J Leedman. Submitted to Cancer Research . The relative contribution of authors is as follows: Christin Down (the Candidate): 55% Dianne J Beveridge 6 % Michael R Epis 6 % Ricky Lareu 3 % Lisa M Stuart 3 % Britt Granath 3 % Dominic Voon 3 % Henry Furneaux 3 % Cecily Metcalf 3 % Jacqueline Bentel 5 % Peter J Leedman 10 % Data from the manuscript described above are presented in this thesis in the following figures, and were performed by the investigator indicated: Figure 3.1A : Dr Cecily Metcalfe, Department of Pathology, Royal Perth Hospital. -

Supplemental Figure S1 Differentially Methylated Regions (Dmrs
Supplemental Figure S1 '$$#0#,2'**7+#2&7*2#"0#%'-,11 #25##,"'1#1#122#1 '!2-0'*"#.'!2'-,-$122,1'2'-,$0-+2- !"Q !"2-$%," $ 31',% 25-$-*" !&,%# ," ' 0RTRW 1 !32V-$$ !0'2#0'T - #.0#1#,22'-, -$ "'$$#0#,2'**7+#2&7*2#"%#,#11',.0#,2#1,"2&#'0 #&4'-022,1'2'-, #25##,"'$$#0#,2"'1#1#122#1T-*!)00-51',"'!2#&7.#0+#2&7*2#"%#,#1Q%0700-51 &7.-+#2&7*2#"%#,#1Q31',%25-$-*"!&,%#,"'0RTRW1!32V-$$!0'2#0'T-%#,#1 +#22&# -4#!0'2#0'22,1'2'-,$0-+$%2-$Q5#2�#$-0#*1-',!*3"#" %#,#15'2&V4*3#0RTRWT$$#!2#"%#,10#&'%&*'%&2#" 712#0'1)1#T Supplemental Figure S2 Validation of results from the HELP assay using Epityper MassarrayT #13*21 $0-+ 2&# 1$ 117 5#0# !-00#*2#" 5'2& /3,2'22'4# +#2&7*2'-, ,*78#" 7 '13*$'2#11007$-04V-,"6U-%#,#.0-+-2#00#%'-,1T11007 51.#0$-0+#"31',%**4'* *#1+.*#1T S Supplemental Fig. S1 A unique hypermethylated genes (methylation sites) 454 (481) 5693 (6747) 120 (122) NLMGUS NEWMM REL 2963 (3207) 1338 (1560) 5 (5) unique hypomethylated genes (methylation sites) B NEWMM 0 (0) MGUS 454 (481) 0 (0) NEWMM REL NL 3* (2) 2472 (3066) NEWMM 2963 REL (3207) 2* (2) MGUS 0 (0) REL 2 (2) NEWMM 0 (0) REL Supplemental Fig. S2 A B ARID4B DNMT3A Methylation by MassArray Methylation by MassArray 0 0.2 0.4 0.6 0.8 1 1.2 0.5 0.6 0.7 0.8 0.9 1 2 0 NL PC MGUS 1.5 -0.5 NEW MM 1 REL MM -1 0.5 -1.5 0 -2 -0.5 -1 -2.5 -1.5 -3 Methylation by HELP Assay Methylation by HELP Methylation by HELP Assay Methylation by HELP -2 -3.5 -2.5 -4 Supplemental tables "3..*#+#,2*6 *#"SS 9*','!*!&0!2#0'12'!1-$.2'#,21+.*#1 DZ_STAGE Age Gender Ethnicity MM isotype PCLI Cytogenetics -

Improving the Yeast Two-Hybrid System with Permutated Fusion Proteins: the Varicella Zoster Virus Protein Interaction Network
Improving the Yeast two-hybrid system with permutated fusion proteins: The Varicella zoster virus protein interaction network Zur Erlangung des akademischen Grades eines DOKTORS DER NATURWISSENSCHAFTEN (Dr. rer. nat.) Fakultät für Chemie und Biowissenschaften Karlsruher Institut für Technologie (KIT) - Universitätsbereich vorgelegte DISSERTATION von Diplom-Biologe Thorsten Stellberger aus Rauenberg Dekan: Prof. Dr. Stefan Bräse Referent: Prof. Dr. Jonathan Sleeman Korreferent: Prof. Dr. Jörg Kämper Tag der mündlichen Prüfung: 20.10.2010 Die vorliegende Arbeit wurde in der Zeit von Januar 2007 bis September 2010 in der Arbeitsgruppe von PD Dr. Peter Uetz im Institut für Toxikologie und Genetik des Karlsruher Instituts für Technologie (KIT), Campus Nord, angefertigt. Zusammenfassung Die vorliegende Studie beschreibt den ersten Versuch zur Erstellung eines vergleichenden Protein-Protein Interaktionsnetzwerks mit dem Yeast two-hybrid (Y2H) System. Viele Studien bestätigen, dass Interaktionsnetzwerke aus proteomweiten Screens unvollständig sind. Dies stützt sich auf die Beobachtung, dass Interaktionsdaten, die mit unterschiedlichen Methoden erhoben wurden, nur geringe Überlappungen zeigen. Dies betrifft auch unterschiedliche Systeme innerhalb einer Methode, wie verschiedene Y2H-Systeme. Dadurch wurde die Frage aufgeworfen, welche Rolle strukturelle Unterschiede, insbesondere sterische Bedingungen im Testsystem spielen, verursacht durch die Orientierung der verwendeten Fusionsdomänen. In dieser Arbeit untersuche ich deren Einfluss auf die Detektierbarkeit -
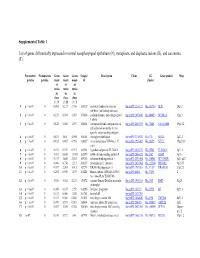
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Anti-Human ZMYM3, Monoclonal Alternate Names: Zinc Finger MYM-Type Protein 3
1 of 5 Anti-Human ZMYM3, monoclonal Alternate Names: Zinc finger MYM-type protein 3 Cat. No. 15-026 FOR RESEARCH USE ONLY NOT FOR USE IN HUMANS PRODUCT DESCRIPTION UniProt Summary Physical Characteristics UniProt Quantity: 1 ml (culture supernatant) or Specificity: monospecific for human Primary Accession: Q14202 100ug at 1mg/ml (purified) ZMYM3 ; see “Microarray Analysis” below Plays a role in the regulation of cell morphology and Format: culture supernatant or purified cytoskeletal organization. May be a component of a material Reactivity: human; not tested for BHC histone deacetylase complex that contains cross reactivity in other species HDAC1, HDAC2, HMG20B/BRAF35, KDM1A, Host/Isotype: mouse IgG2a RCOR1/CoREST, PHF21A/BHC80, ZMYM2, Stability/Storage: 12 months long ZNF217, ZMYM3, GSE1 and GTF2I. Nucleus (By Clonality: monoclonal; ID term: -20°C; short term: 4°C; avoid similarity). Event=Alternative splicing; Named freeze-thaw cycles; aliquot as required isoforms=3; Name=1; IsoId=Q14202-1; JH39.2.2F10 Sequence=Displayed; Name=2; IsoId=Q14202-2; Sequence=VSP_004492; Note=No experimental Formulation: culture supernatant Handling Notes: small volumes of confirmation available; Name=3; IsoId=Q14202-3; contains 0.02% NaN3. Purified material antibody may occasionally become Sequence=VSP_043262, VSP_043263; Most contains 30% glycerol, PBS and 0.02% entrapped in the seal of the product vial abundant in brain, moderate in muscle and heart, NaN3 during shipment and storage; briefly low in other tissues except placenta. Note=A centrifuge the vial on a tabletop chromosomal aberration involving ZMYM3 may be a centrifuge to dislodge any liquid in the cause of X-linked mental retardation.. -

Supplementary Table 1: Significantly
Supplementary Table 1: Significantly upregulated genes in the “malignant” cluster of PNETs (p<0.05) Fold change Gene (WDEC/WDET) FEV protein 11.61 adenylate cyclase 2 (brain) 4.47 nuclear receptor subfamily 4, group A, member 2 4.45 growth arrest and DNA-damage-inducible, beta 3.28 plexin B1 3.10 neuronal PAS domain protein 2 2.92 caldesmon 1 2.81 drebrin 1 2.43 hypothetical protein BC013764 2.41 TGFB-induced factor (TALE family homeobox) 2.26 Homo sapiens cDNA FLJ12843 fis, clone NT2RP2003293, mRNA sequence 2.17 nuclear prelamin A recognition factor 2.15 chromobox homolog 4 (Pc class homolog, Drosophila) 2.14 RuvB (E coli homolog)-like 1 [Homo sapiens], mRNA sequence 2.03 latent transforming growth factor beta binding protein 4 2.02 putative glialblastoma cell differentiation-related 1.92 hypothetical protein FLJ10298 1.92 protein tyrosine phosphatase type IVA, member 1 1.90 ADP-ribosylation factor-like 4 1.88 NIMA (never in mitosis gene a)-related kinase 7 1.87 RAB11B, member RAS oncogene family 1.87 myelin transcription factor 1 1.86 centaurin, beta 2 1.83 Homo sapiens cDNA FLJ38575 fis, clone HCHON2007046, mRNA sequence 1.83 SH3 domain binding glutamic acid-rich protein like 3 1.81 immunoglobulin heavy constant gamma 3 (G3m marker) 1.77 chromobox homolog 3 (HP1 gamma homolog, Drosophila) 1.74 basic transcription element binding protein 1 1.73 H1 histone family, member X 1.73 protein phosphatase 2A, regulatory subunit B' (PR 53) 1.73 Homo sapiens cDNA FLJ30096 fis, clone BNGH41000045, mRNA sequence 1.73 KIAA0397 gene product 1.71 OK/SW-CL.4 [Homo sapiens], mRNA sequence 1.69 high-mobility group 20B 1.69 phosphoserine phosphatase 1.68 Homo sapiens mRNA; cDNA DKFZp434B142 (from clone DKFZp434B142), mRNA sequence 1.67 nuclear mitotic apparatus protein 1 1.67 hypothetical protein 628 1.66 PRKC, apoptosis, WT1, regulator 1.64 E3 ubiquitin ligase SMURF1 1.64 chromodomain protein, Y chromosome-like 1.63 hypothetical protein FLJ21839 1.62 hypothetical protein MGC2550 1.62 Suppl. -

Directed Differentiation of Human Embryonic Stem Cells Into Haematopoietic and Definitive Endodermal Lineages
DIRECTED DIFFERENTIATION OF HUMAN EMBRYONIC STEM CELLS INTO HAEMATOPOIETIC AND DEFINITIVE ENDODERMAL LINEAGES ABRAHAM SUMAN MARY (M.Sc MICROBIOLOGY, UNIV. OF MUMBAI, INDIA) A THESIS SUBMITTED FOR THE DEGREE OF MASTER OF SCIENCE DEPARTMENT OF BIOCHEMISTRY NATIONAL UNIVERSITY OF SINGAPORE 2009 ACKNOWLEDGEMENTS In all things I give YOU glory! You have always led me through amazing paths and given me gifts that I don’t deserve. I thank you Lord for all the blessings you constantly shower on me. Everything is possible with God! I thank Dr. Alan Colman for being my guide and helping me to initiate the work contained in this dissertation. His support and encouragement have been invaluable. Thank you, Alan for your support through the years. Dr. Norris Ray Dunn took me under his wing and guided me through this endeavour. He has been a true mentor, always willing to teach, and I have learned a lot from him. Ray, thank you for showing me the way and helping me reach this juncture. A significant part of the research was conducted at ES Cell International Pte Ltd to whom I would like to express my sincere gratitude. Triona, Jacqui, Robert, Michael, Bruce, Chirag, Suzan and so many others have played important roles and encouraged me at all times. Critical portions of this work were done at the Institute of Medical Biology, A*Star. I would like to register my appreciation for the support and help provided by many people in IMB especially, members of the Ray Dunn lab, Mike Jones lab and Alan Colman lab. Kee Yew, thank you for giving me your precious time and helping me with some of the most important data in this dissertation. -

Supplementary Table B
Table B: Liposarcoma weighted gene analysis Group Weight p Value Clone ID Gene Name Clone Title group 1 12.121071 0 774036 GSTTLp28 glutathione-S-transferase like; glutathione transferase omega group 1 11.840005 0 470128 MYO1E myosin IE group 1 11.065529 0 754582 EVI2A ecotropic viral integration site 2A group 1 10.086113 0 1499940 MAP1A microtubule-associated protein 1A group 1 10.060539 0 271952 ARL7 ADP-ribosylation factor-like 7 group 1 9.942134 0 47481 CLECSF2 C-type (calcium dependent, carbohydrate-recognition domain) lectin, superfamily member 2 group 1 9.768436 0 898305 NBL1 neuroblastoma, suppression of tumorigenicity 1 group 1 9.513434 0 506548 RGS10 regulator of G-protein signalling 10 group 1 9.19263 0 714106 PLAU plasminogen activator, urokinase group 1 9.112408 0 813841 PLAT plasminogen activator, tissue group 1 9.060205 0 139009 FN1 fibronectin 1 group 1 8.980438 0 73852 Homo sapiens cDNA FLJ14388 fis, clone HEMBA1002716 group 1 8.797709 0 823925 CKIP-1 CK2 interacting protein 1; HQ0024c protein group 1 8.76157 0 810224 DKFZp564I1922 DKFZP564I1922 protein group 1 8.583343 0 502603 RAB23 RAB23, member RAS oncogene family group 1 8.531602 0 700299 WASPIP Wiskott-Aldrich syndrome protein interacting protein group 1 8.530959 0 627248 SBBI31 SBBI31 protein group 1 8.524575 0 174627 SCG2 secretogranin II (chromogranin C) group 1 8.524259 0 1031203 NCYM DNA-binding transcriptional activator group 1 8.483101 0 1606557 FHL2 four and a half LIM domains 2 group 1 8.414318 0 231675 EVI2A ecotropic viral integration site 2A group 1 8.390458