Born in Braunschweig
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table 1 Genes Tested in Qrt-PCR in Nfpas
Supplementary Table 1 Genes tested in qRT-PCR in NFPAs Gene Bank accession Gene Description number ABI assay ID a disintegrin-like and metalloprotease with thrombospondin type 1 motif 7 ADAMTS7 NM_014272.3 Hs00276223_m1 Rho guanine nucleotide exchange factor (GEF) 3 ARHGEF3 NM_019555.1 Hs00219609_m1 BCL2-associated X protein BAX NM_004324 House design Bcl-2 binding component 3 BBC3 NM_014417.2 Hs00248075_m1 B-cell CLL/lymphoma 2 BCL2 NM_000633 House design Bone morphogenetic protein 7 BMP7 NM_001719.1 Hs00233476_m1 CCAAT/enhancer binding protein (C/EBP), alpha CEBPA NM_004364.2 Hs00269972_s1 coxsackie virus and adenovirus receptor CXADR NM_001338.3 Hs00154661_m1 Homo sapiens Dicer1, Dcr-1 homolog (Drosophila) (DICER1) DICER1 NM_177438.1 Hs00229023_m1 Homo sapiens dystonin DST NM_015548.2 Hs00156137_m1 fms-related tyrosine kinase 3 FLT3 NM_004119.1 Hs00174690_m1 glutamate receptor, ionotropic, N-methyl D-aspartate 1 GRIN1 NM_000832.4 Hs00609557_m1 high-mobility group box 1 HMGB1 NM_002128.3 Hs01923466_g1 heterogeneous nuclear ribonucleoprotein U HNRPU NM_004501.3 Hs00244919_m1 insulin-like growth factor binding protein 5 IGFBP5 NM_000599.2 Hs00181213_m1 latent transforming growth factor beta binding protein 4 LTBP4 NM_001042544.1 Hs00186025_m1 microtubule-associated protein 1 light chain 3 beta MAP1LC3B NM_022818.3 Hs00797944_s1 matrix metallopeptidase 17 MMP17 NM_016155.4 Hs01108847_m1 myosin VA MYO5A NM_000259.1 Hs00165309_m1 Homo sapiens nuclear factor (erythroid-derived 2)-like 1 NFE2L1 NM_003204.1 Hs00231457_m1 oxoglutarate (alpha-ketoglutarate) -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Product Information
Product information PLDN, 1-172aa Human, His-tagged, Recombinant, E.coli Cat. No. IBATGP1472 Full name: pallidin NCBI Accession No.: NP_036520 Synonyms: HPS9, PA, PALLID Description: PLDN (Pallidin) may play a role in intracellular vesicle trafficking. It interacts with Syntaxin 13 which mediates intracellular membrane fusion. Several alternatively spliced transcript variants of this gene have been described, but the full-length nature of some of these variants has not been determined. This protein involved in the development of lysosome-related organelles, such as melanosomes and platelet-dense granules. PLDN has been shown to interact with BLOC1S1, STX12, Dysbindin, CNO, BLOC1S2, MUTED and SNAPAP. Recombinant human PLDN protein, fused to His-tag at N-terminus, was expressed in E.coli and purified by using conventional chromatography techniques. Form: Liquid. In 20mM Tris-HCl buffer (pH 8.0) containing 2mM DTT, 10% glycerol, 100mM NaCl Molecular Weight: 21.9kDa (192aa), confirmed by MALDI-TOF (Molecular weight on SDS-PAGE will appear higher) Purity: > 90% by SDS - PAGE Concentration: 1mg/ml (determined by Bradford assay) 15% SDS-PAGE (3ug) Sequences of amino acids: MGSSHHHHHH SSGLVPRGSH MSVPGPSSPD GALTRPPYCL EAGEPTPGLS DTSPDEGLIE DLTIEDKAVE QLAEGLLSHY LPDLQRSKQA LQELTQNQVV LLDTLEQEIS KFKECHSMLD INALFAEAKH YHAKLVNIRK EMLMLHEKTS KLKKRALKLQ QKRQKEELER EQQREKEFER EKQLTARPAK RM General references: Huang L, et al. (1999). Nat Genet 23 (3): 329–32. Moriyama K., et al. (2002) Traffic 3:666-677 Storage: Can be stored at +4°C short term (1-2 weeks). For long term storage, aliquot and store at -20°C or -70°C. Avoid repeated freezing and thawing cycles. For research use only. This product is not intended or approved for human, diagnostics or veterinary use. -

SNAPAP Antibody Cat
SNAPAP Antibody Cat. No.: XW-8015 SNAPAP Antibody Specifications HOST SPECIES: Chicken SPECIES REACTIVITY: Human, Mouse, Rat IMMUNOGEN: 31-136 TESTED APPLICATIONS: WB SNARE-associated protein Snapin antibody can be used for the detection of SNARE- APPLICATIONS: associated protein Snapin by Western Blot. PREDICTED MOLECULAR 14.9 kDa (calculated) WEIGHT: Properties PURIFICATION: Immunoaffinity Purified CLONALITY: Polyclonal CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: Phosphate-Buffered Saline. No preservatives added. CONCENTRATION: 1 mg/mL October 1, 2021 1 https://www.prosci-inc.com/snapap-antibody-8015.html SNAPAP antibody can be stored at 4˚C for short term (weeks). Long term storage should STORAGE CONDITIONS: be at -20˚C. As with all antibodies care should be taken to avoid repeated freeze thaw cycles. Antibodies should not be exposed to prolonged high temperatures. Additional Info OFFICIAL SYMBOL: SNAPIN BLOC1S7, SNAP25BP, SNAPAP, SNARE-associated protein Snapin, Biogenesis of lysosome- related organelles complex 1 subunit 7, BLOC-1 subunit 7, BLOS7, BLOC1S7, ALTERNATE NAMES: SNAREassociated protein Snapin, Synaptosomal-associated protein 25 binding protein, SNAP-associated protein ACCESSION NO.: NP_036569.1 PROTEIN GI NO.: 6912674 GENE ID: 23557 USER NOTE: Optimal dilutions for each application to be determined by the researcher. Background and References FUNCTION: May modulate a step between vesicle priming, fusion and calcium-dependent neurotransmitter release by potentiating the interaction of synaptotagmins with the SNAREs and the plasma-membrane-associated protein SNAP25. Its phosphorylation state BACKGROUND: influences exocytotic protein interactions and may regulate synaptic vesicle exocytosis. May also have a role in the mechanisms of SNARE-mediated membrane fusion in non- neuronal cells (By similarity). -
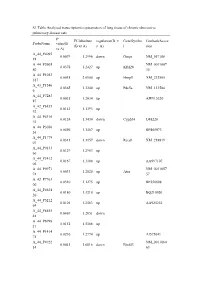
S1 Table Analyzed Transcriptomics Parameters of Lung Tissue of Chronic
S1 Table Analyzed transcriptomics parameters of lung tissue of chronic obstructive pulmonary disease rats p- FCAbsolute regulation(B_v GeneSymbo GenbankAcces ProbeName value(B (B vs A) s_A) l sion vs A) A_44_P4095 0.0079 1.2996 down Gmpr NM_057188 18 A_44_P2605 NM_0011067 0.0378 1.2427 up Klhl28 80 35 A_44_P1052 0.0018 2.0900 up Hnrpll XM_233805 387 A_43_P1546 0.0365 1.3240 up Pde5a NM_133584 6 A_44_P7283 0.0035 1.2634 up AW915320 87 A_42_P8435 0.0143 1.1393 up 92 A_44_P4310 0.0124 1.3434 down Cyp2d4 U48220 92 A_44_P3056 0.0496 1.1467 up BP465973 36 A_44_P1779 0.0311 1.1157 down Rccd1 XM_218819 07 A_44_P9133 0.0129 1.2963 up 60 A_44_P3412 0.0167 1.3108 up AA997107 04 A_44_P9971 NM_0011057 0.0033 1.2820 up Atrx 74 57 A_42_P7763 0.0310 1.1275 up BF556698 00 A_44_P4634 0.0180 1.3210 up BQ210920 20 A_44_P5212 0.0101 1.2583 up AA925252 64 A_44_P8855 0.0469 1.2051 down 48 A_44_P6998 0.0151 1.5168 up 21 A_44_P1414 0.0236 1.2774 up AI575641 75 A_44_P4922 NM_0011064 0.0041 1.6816 down Eps8l3 14 63 A_43_P2202 0.0488 1.2176 up Setx XM_342400 0 A_44_P2066 0.0205 1.2767 up BF417274 31 A_44_P6213 0.0007 2.0237 up 04 A_44_P3775 0.0068 1.2952 up 20 A_44_P6058 0.0365 1.1240 up 38 A_44_P2861 0.0022 1.4896 up XM_226020 58 A_44_P9139 0.0108 1.5661 up 62 A_43_P1214 0.0058 1.9100 up Hnf4a NM_022180 6 A_44_P6525 NM_0011300 0.0036 1.1586 down Kdm1 85 98 A_44_P3541 0.0061 1.3319 down Cep250 XR_009560 50 A_44_P5160 RGD131055 NM_0011068 0.0293 1.2854 down 73 2 38 A_44_P2237 0.0192 1.3087 up AW142694 41 A_43_P1552 0.0096 1.2036 up Ccnd3 NM_012766 4 A_44_P4098 0.0479 -

Downregulation of Carnitine Acyl-Carnitine Translocase by Mirnas
Page 1 of 288 Diabetes 1 Downregulation of Carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion Mufaddal S. Soni1, Mary E. Rabaglia1, Sushant Bhatnagar1, Jin Shang2, Olga Ilkayeva3, Randall Mynatt4, Yun-Ping Zhou2, Eric E. Schadt6, Nancy A.Thornberry2, Deborah M. Muoio5, Mark P. Keller1 and Alan D. Attie1 From the 1Department of Biochemistry, University of Wisconsin, Madison, Wisconsin; 2Department of Metabolic Disorders-Diabetes, Merck Research Laboratories, Rahway, New Jersey; 3Sarah W. Stedman Nutrition and Metabolism Center, Duke Institute of Molecular Physiology, 5Departments of Medicine and Pharmacology and Cancer Biology, Durham, North Carolina. 4Pennington Biomedical Research Center, Louisiana State University system, Baton Rouge, Louisiana; 6Institute for Genomics and Multiscale Biology, Mount Sinai School of Medicine, New York, New York. Corresponding author Alan D. Attie, 543A Biochemistry Addition, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin, (608) 262-1372 (Ph), (608) 263-9608 (fax), [email protected]. Running Title: Fatty acyl-carnitines enhance insulin secretion Abstract word count: 163 Main text Word count: 3960 Number of tables: 0 Number of figures: 5 Diabetes Publish Ahead of Print, published online June 26, 2014 Diabetes Page 2 of 288 2 ABSTRACT We previously demonstrated that micro-RNAs 132 and 212 are differentially upregulated in response to obesity in two mouse strains that differ in their susceptibility to obesity-induced diabetes. Here we show the overexpression of micro-RNAs 132 and 212 enhances insulin secretion (IS) in response to glucose and other secretagogues including non-fuel stimuli. We determined that carnitine acyl-carnitine translocase (CACT, Slc25a20) is a direct target of these miRNAs. -

As a Model for Lysosomal Storage Disorders Gert De Voer, Dorien Peters, Peter E.M
as a model for lysosomal storage disorders Gert de Voer, Dorien Peters, Peter E.M. Taschner To cite this version: Gert de Voer, Dorien Peters, Peter E.M. Taschner. as a model for lysosomal storage disorders. Biochimica et Biophysica Acta - Molecular Basis of Disease, Elsevier, 2008, 1782 (7-8), pp.433. 10.1016/j.bbadis.2008.04.003. hal-00501575 HAL Id: hal-00501575 https://hal.archives-ouvertes.fr/hal-00501575 Submitted on 12 Jul 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ÔØ ÅÒÙ×Ö ÔØ Caenorhabditis elegans as a model for lysosomal storage disorders Gert de Voer, Dorien Peters, Peter E.M. Taschner PII: S0925-4439(08)00093-8 DOI: doi: 10.1016/j.bbadis.2008.04.003 Reference: BBADIS 62810 To appear in: BBA - Molecular Basis of Disease Received date: 13 May 2007 Revised date: 23 April 2008 Accepted date: 24 April 2008 Please cite this article as: Gert de Voer, Dorien Peters, Peter E.M. Taschner, Caenorhab- ditis elegans as a model for lysosomal storage disorders, BBA - Molecular Basis of Disease (2008), doi: 10.1016/j.bbadis.2008.04.003 This is a PDF file of an unedited manuscript that has been accepted for publication. -

NIH Public Access Author Manuscript Annu Rev Genomics Hum Genet
NIH Public Access Author Manuscript Annu Rev Genomics Hum Genet. Author manuscript; available in PMC 2009 October 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Annu Rev Genomics Hum Genet. 2008 ; 9: 359±386. doi:10.1146/annurev.genom.9.081307.164303. Disorders of Lysosome-related Organelle Biogenesis: Clinical and Molecular Genetics Marjan Huizing1, Amanda Helip-Wooley2, Wendy Westbroek2, Meral Gunay-Aygun2, and William A. Gahl2 Marjan Huizing: [email protected]; Amanda Helip-Wooley: [email protected]; Wendy Westbroek: [email protected]; Meral Gunay-Aygun: [email protected]; William A. Gahl: [email protected] 1 Cell Biology of Metabolic Disorders Unit, National Institutes of Health, Bethesda, Maryland 20892 2 Section on Human Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892 Abstract Lysosome-related organelles (LROs) are a heterogeneous group of vesicles that share various features with lysosomes, but are distinct in function, morphology, and composition. The biogenesis of LROs employs a common machinery, and genetic defects in this machinery can affect all LROs or only an individual LRO, resulting in a variety of clinical features. In this review, we discuss the main components in LRO biogenesis. We also address the function, composition and resident cell type of the major LROs. Finally, we describe the clinical characteristics of the major human LRO disorders. Keywords Chediak-Higashi syndrome; Griscelli syndrome; Hermansky-Pudlak syndrome; melanosome; platelet INTRODUCTION Lysosomes are membrane-bound cytoplasmic organelles that serve as major degradative compartments in eukaryotic cells (65). -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name SNAP-associated protein Gene Symbol SNAPIN Organism Human Gene Summary SNAPAP is a component of the SNARE complex of proteins that is required for synaptic vesicle docking and fusion (Ilardi et al. 1999 Gene Aliases SNAPAP RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.32018 Ensembl Gene ID ENSG00000143553 Entrez Gene ID 23557 Assay Information Unique Assay ID qHsaCID0017371 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000368685 Amplicon Context Sequence AACTGTGCCGCATAAATGAGGATCAGAAGGTGGCCCTGGATCTTGACCCCTATG TTAAGAAGCTACTTAATGCCCGGCGACGCGTTGTCTTGGTTAACAACATTCTACA GAATGCTCAGGAACGACTGAGACGGCTAAACCACAGTGTTGCCAAGGAAACAGC CCGCAGGAGAGCAATG Amplicon Length (bp) 149 Chromosome Location 1:153631924-153633735 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9998 cDNA Cq 20.86 cDNA Tm (Celsius) 84 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report SNAPIN, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Neurodevelopmental Disease Mechanisms, Primary Cilia, and Endosomes Converge on the BLOC-1 and BORC Complexes Cortnie Hartwig, Emory University William J
Neurodevelopmental disease mechanisms, primary cilia, and endosomes converge on the BLOC-1 and BORC complexes Cortnie Hartwig, Emory University William J. Monis, University of Massachusetts Xun Chen, University of Southern California Dion K. Dickman, University of Southern California Gregory J. Pazour, University of Massachusetts Victor Faundez, Emory University Journal Title: Developmental Neurobiology Volume: Volume 78, Number 3 Publisher: Wiley: 12 months | 2018-03-01, Pages 311-330 Type of Work: Article | Post-print: After Peer Review Publisher DOI: 10.1002/dneu.22542 Permanent URL: https://pid.emory.edu/ark:/25593/tnvw9 Final published version: http://dx.doi.org/10.1002/dneu.22542 Copyright information: © 2017 Wiley Periodicals, Inc. Accessed September 29, 2021 9:42 PM EDT HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Dev Neurobiol Manuscript Author . Author manuscript; Manuscript Author available in PMC 2019 March 01. Published in final edited form as: Dev Neurobiol. 2018 March ; 78(3): 311–330. doi:10.1002/dneu.22542. Neurodevelopmental Disease Mechanisms, Primary Cilia, and Endosomes Converge on the BLOC-1 and BORC Complexes Cortnie Hartwig1, William J. Monis2, Xun Chen3, Dion K. Dickman3, Gregory J. Pazour2, and Victor Faundez1,# 1Department of Cell Biology. Emory University Atlanta, Georgia 30322 2Program in Molecular Medicine, University of Massachusetts Medical School, Biotech II, Worcester, MA 01605 3Department of Biology, Neurobiology Section, University of Southern California, Los Angeles, California 90089 Abstract The Biogenesis of Lysosome-related Organelles Complex-1 (BLOC-1) and the BLOC-One- Related Complex (BORC) are cytosolic protein complexes required for specialized membrane protein traffic along the endocytic route and for the spatial distribution of endosome-derived compartments, respectively. -

Data Sheet 148
Rudolf-Wissell-Str. 28a Background 37079 Göttingen, Germany Phone: +49 551-50556-0 Snapin, also referred to as Snapap, was initially identified as a SNAP 25 interacting protein which Fax: +49 551-50556-384 enhances the binding of synaptotagmin 1 to SNAREs in a phosphorylation dependent manner. Later E-mail: [email protected] an ubiquitous expression pattern in neuronal and non-neuronal cells and interaction with SNAP 23 Web: www.sysy.com was described. The protein contains heptad repeats typical for coiled coils in its C-terminal part. Snapin The role of this protein in SNARE mediated fusion is still under discussion. Cat.No. 148 002; Polyclonal rabbit antibody, 200 µl antiserum (lyophilized) Selected References for 148 002 Snapin, positive regulator of stimulation- induced Ca²⁺ release through RyR, is necessary for HIV-1 replication in T cells. Data Sheet Kinoshita SM, Kogure A, Taguchi S, Nolan GP PloS one (2013) 810: e75297. WB, IP, ICC; tested species: human Reconstitution/ 200 µl antiserum, lyophilized. For reconstitution add 200 µl H2O, then aliquot and The SNARE-associated component SNAPIN binds PUMILIO2 and NANOS1 proteins in human male germ cells. Storage store at -20°C until use. Ginter-Matuszewska B, Spik A, Rembiszewska A, Koyias C, Kupryjanczyk J, Jaruzelska J For detailed information, see back of the data sheet. Molecular human reproduction (2009) 153: 173-9. WB, IHC-P; tested species: human,mouse Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to Applications WB: 1 : 100 up to 1 : 5000 mycobacteria. -

Supplementary Figure S1. Generation of Floxed Nphs2 Exon 2 Allele and Confirmation
CONDITIONAL PODOCIN INACTIVATION SUPPLEMENTARY DATA SUPPLEMENTARY FIGURES AND TABLES: Supplementary Figure S1. Generation of floxed Nphs2 exon 2 allele and confirmation of gene targeting. A. A 7.7-kb targeting vector consisting of a loxP site in intron 1-2 and a loxP-Frt-Neo-Frt cassette in intron 2-3 was generated by PCR and inserted into an MCI proprietary vector. The linearized vector was electroporated into ES cells. B. Screening of 400 ES cell clones by Southern blot hybridization, using a Neo probe and a 3' probe, revealed two correctly targeted clones 121 and 130. A Target vector 1 2 NEO 5’ end 3’ end 3’ probe Wild-type allele 1 2 3 9.2 kb Xmn I Xmn I 5’ end neo probe Floxed allele 1 2 NEO 3 5 kb Xmn I Xmn I 7.8 kb Nde I Nde I B Neo probe, NdeI 3’ probe, XmnI WT 121 130 121 130 MW MW - 1 - Supplementary Figure S2. Generation of Nphs2lox2/-,Cre+ mice and excision of exon 2 upon Cre recombinase induction. A. Triallelic Nphs2lox2/-,Cre+ mice were obtained by mating phenotypically normal Nphs2lox2/lox2 mice with Nphs2+/-,Cre+ mice. Mendelian inheritance of these alleles was observed. B. Genotypes were verified by multiplex PCR of tail genomic DNA. C. Cre recombinase activity was induced upon tamoxifen administration, leading to excision of the floxed exon 2 of the Nphs2 gene. D. Cre recombinase activity in the kidney was verified by PCR using a set of forward and reverse primers designed around exon 2 and demonstrating a 692-bp product (before Cre) and a 316-bp product (after Cre) using genomic DNA extracted from the renal cortex.