Occupational Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

USMLE – What's It
Purpose of this handout Congratulations on making it to Year 2 of medical school! You are that much closer to having your Doctor of Medicine degree. If you want to PRACTICE medicine, however, you have to be licensed, and in order to be licensed you must first pass all four United States Medical Licensing Exams. This book is intended as a starting point in your preparation for getting past the first hurdle, Step 1. It contains study tips, suggestions, resources, and advice. Please remember, however, that no single approach to studying is right for everyone. USMLE – What is it for? In order to become a licensed physician in the United States, individuals must pass a series of examinations conducted by the National Board of Medical Examiners (NBME). These examinations are the United States Medical Licensing Examinations, or USMLE. Currently there are four separate exams which must be passed in order to be eligible for medical licensure: Step 1, usually taken after the completion of the second year of medical school; Step 2 Clinical Knowledge (CK), this is usually taken by December 31st of Year 4 Step 2 Clinical Skills (CS), this is usually be taken by December 31st of Year 4 Step 3, typically taken during the first (intern) year of post graduate training. Requirements other than passing all of the above mentioned steps for licensure in each state are set by each state’s medical licensing board. For example, each state board determines the maximum number of times that a person may take each Step exam and still remain eligible for licensure. -

Statin Myopathy: a Common Dilemma Not Reflected in Clinical Trials
REVIEW CME EDUCATIONAL OBJECTIVE: Readers will assess possible statin-induced myopathy in their patients on statins CREDIT GENARO FERNANDEZ, MD ERICA S. SPATZ, MD CHARLES JABLECKI, MD PAUL S. PHILLIPS, MD Internal Medicine Residency Program, Robert Wood Johnson Clinical Scholars Department of Neurosciences, University Director, Interventional Cardiology, The University of Utah, Salt Lake City Program, Cardiovascular Disease Fellow, of California San Diego, La Jolla Department of Cardiology, Scripps Mercy Yale University School of Medicine, New Hospital, San Diego, CA Haven, CT Statin myopathy: A common dilemma not reflected in clinical trials ■■ ABSTRACT hen a patient taking a statin complains Wof muscle aches, is he or she experiencing Although statins are remarkably effective, they are still statin-induced myopathy or some other prob- underprescribed because of concerns about muscle toxic- lem? Should statin therapy be discontinued? Statins have proven efficacy in preventing ity. We review the aspects of statin myopathy that are 1 important to the primary care physician and provide a heart attacks and death, and they are the most guide for evaluating patients on statins who present with widely prescribed drugs worldwide. Neverthe- less, they remain underused, with only 50% of muscle complaints. We outline the differential diagnosis, those who would benefit from being on a statin the risks and benefits of statin therapy in patients with receiving one.2,3 In addition, at least 25% of possible toxicity, and the subsequent treatment options. adults who start taking statins stop taking them 4 ■■ by 6 months, and up to 60% stop by 2 years. KEY POINTS Patient and physician fears about myopathy There is little consensus on the definition of statin-in- remain a key reason for stopping. -

SUPPLEMENTARY MATERIAL Supplementary 1. International
SUPPLEMENTARY MATERIAL Supplementary 1. International Myositis Classification Criteria Project Steering Committee Supplementary 2. Pilot study Supplementary 3. International Myositis Classification Criteria Project questionnaire Supplementary 4. Glossary and definitions for the International Myositis Classification Criteria Project questionnaire Supplementary 5. Adult comparator cases in the International Myositis Classification Criteria Project dataset Supplementary 6. Juvenile comparator cases in the International Myositis Classification Criteria Project dataset Supplementary 7. Validation cohort from the Euromyositis register Supplementary 8. Validation cohort from the Juvenile dermatomyositis cohort biomarker study and repository (UK and Ireland) 1 Supplementary 1. International Myositis Classification Criteria Project Steering Committee Name Affiliation Lars Alfredsson Institute for Environmental Medicine, Karolinska Institutet, Stockholm, Sweden Anthony A Amato Department of Neurology, Brigham and Women’s Hospital, Harvard Medical School, Boston, USA Richard J Barohn Department of Neurology, University of Kansas Medical Center, Kansas City, USA Matteo Bottai Institute for Environmental Medicine, Karolinska Institutet, Stockholm, Sweden Matthew H Liang Division of Rheumatology, Immunology and Allergy, Brigham and Women´s Hospital, Boston, USA Ingrid E Lundberg (Project Director) Rheumatology Unit, Department of Medicine, Karolinska University Hospital, Solna, Karolinska Institutet, Stockholm, Sweden Frederick W Miller Environmental -

EM Guidemap - Myopathy and Myoglobulinuria
myopathy EM guidemap - Myopathy and myoglobulinuria Click on any of the headings or subheadings to rapidly navigate to the relevant section of the guidemap Introduction General principles ● endocrine myopathy ● toxic myopathy ● periodic paralyses ● myoglobinuria Introduction - this short guidemap supplements the neuromuscular weakness guidemap and offers the reader supplementary information on myopathies, and a short section on myoglobulinuria - this guidemap only consists of a few brief checklists of "causes of the different types of myopathy" that an emergency physician may encounter in clinical practice when dealing with a patient with acute/subacute muscular weakness General principles - a myopathy is suggested when generalized muscle weakness involves large proximal muscle groups, especially around the shoulder and proximal girdle, and when the diffuse muscle weakness is associated with normal tendon reflexes and no sensory findings - a simple classification of myopathy:- Hereditary ● muscular dystrophies ● congenital myopathies http://www.homestead.com/emguidemaps/files/myopathy.html (1 of 13)8/20/2004 5:14:27 PM myopathy ● myotonias ● channelopathies (periodic paralysis syndromes) ● metabolic myopathies ● mitochondrial myopathies Acquired ● inflammatory myopathy ● endocrine myopathies ● drug-induced/toxic myopathies ● myopathy associated with systemic illness - a myopathy can present with fixed weakness (muscular dystrophy, inflammatory myopathy) or episodic weakness (periodic paralysis due to a channelopathy, metabolic myopathy -

Connective Tissue 5.2.04
Other Connective Tissue Diseases Chester V. Oddis, MD Thomas A. Medsger, Jr, MD Arthur Weinstein, MD Contents 1. Undifferentiated Connective Tissue Disease 2. Idiopathic Inflammatory Myopathy 3. Scleroderma 4. Sjögren’s Syndrome 5. References OTHER CONNECTIVE TISSUE DISEASES 1 1. Undifferentiated Connective Tissue Disease Table 1 The American College of Rheumatology (ACR) has published criteria for several different diseases Clinical Features and Autoantibody Findings commonly referred to as connective tissue disease Possibly Specific for a Defined CTD (CTD). The primary aim of such classification crite - ria is to ensure the comparability among CTD stud - ies in the scientific community. These diseases Clinical Feature include rheumatoid arthritis (RA), systemic sclero - sis (SSc), systemic lupus erythematosus (SLE), Malar rash polymyositis (PM), dermatomyositis (DM), and Sjögren’s syndrome (SS). These are systemic Subacute cutaneous lupus rheumatic diseases which reflects their inflamma - tory nature and protean clinical manifestations with Sclerodermatous skin changes resultant tissue injury. Although there are unifying immunologic features that pathogenetically tie Heliotrope rash these separate CTDs to each other, the individual disorders often remain clinically and even serologi - Gottron’s papules cally distinct. Immunogenetic data and autoanti - body findings in the different CTDs lend further Erosive arthritis support for their distinctive identity and often serves to subset the individual CTD even further, as seen with the myositis syndromes, SLE and SSc. In other cases, it remains difficult to classify individuals Autoantibody with a combination of signs, symptoms, and labora - tory test results. It is this group of patients that have Anti-dsDNA an “undifferentiated” connective tissue disease (UCTD), or perhaps more accurately, an undifferen - Anti-Sm tiated systemic rheumatic disease. -

Notes on Rheumatology Author: Liz Tatman © Dr R
Notes on Rheumatology Author: Liz Tatman Musculoskeletal (MSK) Problems: Principles of Management MSK symptoms Pain – site, radiation (get radiation distally, more common with more proximal joints e.g. shoulder and knee), quality (shooting with nerve entrapment). Onset. With usage (mechanical), at rest (inflammatory) or at night (if no relation to movement concern about bony mets). Bony pain – well localised, doesn’t radiate, progressive, night predominant, not related to movement, due to fractures, neoplasia, Paget’s disease, osteomalacia, osteomyelitis, osteonecrosis. Referred pain – poorly localised, diffuse, improved by rubbing. Can get neuropathic pain sometime after injury. Stress pain – in tight pack positions, if in all directions suggests synovitis. Stiffness – prolonged early morning and inactivity imply inflammatory cause. Swelling and deformity. Instability Locking – sudden inability to move joint, usually painful but transient, post unlocking discomfort. Affects hinge joints (knee, elbow, TMJ). Mechanical problem e.g. synovium, cartilage, split meniscus. Loss of function, disability and handicap. Systemic illness – night sweats, malaise, fatigue, weight loss suggest inflammatory process. Sleep disturbance. Red flags: progressive, well localised progressive night-predominant pain (mets), red hot joint (sepsis), sequential joint involvement (spreading sepsis). History – need to determine if traumatic or non-traumatic, ask about dominant hand. Trauma – time, place, mechanism (twist, blow, crush), magnitude of force, direction of force, first aid, immediate symptoms and progression, previous injuries. Non-trauma – rule out (RESTIT) Referred pain, Embolus, Sepsis, Tumour, Ischaemia, Thrombosis. Systems enquiry for non MSK causes, associated features, drug toxicity, pre-op assessment and involvement of other organs. Especially general malaise (anaemia), hot and shivery (infection), skin (rashes, psoriasis, Raynaud’s), eyes (iritis, scleritis). -

Path Pulmonary Outline
Pathology Pulmonary ATELECTASIS Neonatal Atelectasis ‐ The lungs of the neonate never inflate, a consequence of congenital defect, premature birth (insufficient surfactant), or other consequence, also called Patchy Atelectasis Adult or Acquired Atelectasis ‐ Collapse of Previously Inflated lung, creating areas of “airless parenchyma” ‐ Produces a well‐perfused but poorly‐ventilated region, predisposing for infection Decreased Volume ‐ Is a reversible disorder (except in the case of contraction) outside pleural space ‐ Resorption Atelectasis o Consequence of complete obstruction without impairment to blood flow Blockage o A decreased lung volume results in a mediastinal shift towards affected lung o Caused by a mucous plug associated with Asthma, Bronchitis, or Aspiration Pneumonia ‐ Compression Atelectasis o Consequence of partially or totally filled pleura with exudate (CHF), tumor, air (pneumothorax), blood (hemothorax), when air pressure threatens the function of lungs and great vessels (tension pneumothorax), or with an extra‐pulmonary mass compressing Something within lung parenchyma. pleural space compressing o Compressed lung tissue cannot expand and is therefore poorly ventilated. parenchyma o Compression pushes lung resulting in a mediastinal shift away from affected lung ‐ Contraction Atelectasis o Fibrotic changes prevent expansion, resulting in reduced lung volume and ventilation Fibrotic o This form is irreversible Changes are Irreversible PULMONARY EDEMA Pulmonary Edema is simply the accumulation of fluid in the alveolar -

数字 Accessory Bronchus 副気管支 Accessory Fissure 副葉間裂
数字 accentuation 亢進 accessory 副の 数字 accessory bronchus 副気管支 accessory fissure 副葉間裂 10-year survival 10年生存 accessory lobe 副肺葉 18F-fluorodeoxy glucose (FDG) 18F-フルオロデオキシグルコース accessory lung 副肺 2,3-diphosphoglycerate (2,3-DPG) 2,3ジフォスフォグリセレート accessory nasal sinus 副鼻腔 201TI (thallium-201) タリウム accessory trachea 副気管 5-fluorouracil(FU) 5-フルオロウラシル acclimation 順化 5-HT3 receptor antagonist 5-HT3レセプター拮抗薬 acclimation 馴化 5-hydroxytryptamine 5-ヒドロオキシトリプタミン acclimatization 気候順応 5-year survival 5年生存 acclimatization 順化 99mTc-macroaggregated albumin (99mTc-MAA) 99mTc標識大 acclimatization 馴化 凝集アルブミン accommodation 順応 accommodation 調節 accommodation to high altitude 高所順(適)応 A ACE polymorphism ACE遺伝子多型 acetone body アセトン体 abdomen 腹部 acetonuria アセトン尿[症] abdominal 腹部[側]の acetylcholine(ACh) アセチルコリン abdominal breathing 腹式呼吸 acetylcholine receptor (AchR, AChR) アセチルコリン受容体(レセプ abdominal cavity 腹腔 ター) abdominal pressure 腹腔内圧 acetylcholinesterase (AchE, AChE) アセチルコリンエステラーゼ abdominal respiration 腹式呼吸 achalasia アカラシア abdominal wall reflex 腹壁反射 achalasia 弛緩不能症 abduction 外転 achalasia [噴門]無弛緩[症] aberrant 走性 achromatocyte (achromocyte) 無血色素[赤]血球 aberrant 迷入性 achromatocyte (achromocyte) 無へモグロビン[赤]血球 aberrant artery 迷入動脈 acid 酸 aberration 迷入 acid 酸性 ablation 剥離 acid base equilibrium 酸塩基平衡 abnormal breath sound(s) 異常呼吸音 acid fast 抗酸性の abortive 早産の acid fast bacillus 抗酸菌 abortive 頓挫性(型) acid-base 酸―塩基 abortive 不全型 acid-base balance 酸塩基平衡 abortive pneumonia 頓挫[性]肺炎 acid-base disturbance 酸塩基平衡異常 abrasion 剥離 acid-base equilibrium 酸塩基平衡 abscess 膿瘍 acid-base regulation 酸塩基調節 absolute -

Keyword Associations
Bare Minimum Keyword Associations USMLE & COMLEX Review Northwestern Medical Review [email protected] Northwestern Medical Review PO Box 22174 Lansing, Michigan 48909 www.northwesternmedicalreview.com Copyright © 2015 Northwestern Medical Review eBookit.com and Northwestern Medical Review Second Edition ISBN 978-0-9960-9340-1 All rights reserved. Written, published, and printed in the United States of America. No part of this book may be used, reproduced, or transmitted in any form or by any means, electronic or mechanical without written permission from its author or Northwestern Medical Review. All photos adapted from fotolia.com Northwestern Medical Review claims no rights to USMLE or COMLEX USMLE ® National Board of Medical Examiners COMLEX ® National Board of Osteopathic Medical Examiners This book is adapted from the first chapter of Primary Care for the USMLE and COMLEX by Northwestern Medical Review. The full book version is primarily intended to accompany online or live review courses from Northwestern Medical Review. How to use this book This book is adapted from the first chapter of Northwestern Medical Review’s Primary Care for the USMLE and COMLEX book. It contains exceptionally high-yield material found on board exams. The mini-chapters are divided into sections containing commonly tested concepts with keywords that should immediately come to mind while you’re taking your exam. In the back of the book are two additional sections: a Test Your Knowledge section containing questions and answers with explanations from our follow-along lecture workbooks and NMR Question Bank, and a Sample Mnemonics section containing examples of memorable mnemonics from our course material. -

General Disease Finder 455
453 General disease finder 455 This overview will help to find neuromuscular disease patterns in the different sections Cushing‘s disease: steroid myopathy Adrenal dysfunction Addison’s disease: general muscle weakness Periodic paralysis Aldosteronism Tetanic muscles CN: VII AIDS Polyneuropathies: inflammatory, immune mediated, treatment related Myopathies: inflammatory, treatment related Neoplastic: Lymphoma (direct invasion) Opportunistic infections: CMV, Toxoplasmosis, Cryptococcus, HSV, Candida, Varicella, Histoplasma, TBC, Aspergillus CMV polyradiculomyelopathy Herpes zoster radiculitis Syphilitic radiculopathy Treatment related: polyneuropathy/myopathy Ddl, ddC, Foscarnet, Isoniazid Zidovudine Polyneuropathy (distal, rarely proximal, rare ulcers) Alcoholism Mononeuropathy-radial nerve (compression) Myopathy Acute necrotizing myopathy and myoglobinuria Chronic proximal weakness Hypokalemic paralysis Myoglobinuria Compartment syndromes (prolonged compression) Familial amyloid polyneuropathies Amyloid Transthyretin Sensorimotor neuropathy Autonomic involvement Apolipoprotein A-1 Polyneuropathy, painful, hearing loss Gelsolin type V, VII and other CN Mild polyneuropathy Primary amyloidosis (AL) Deposition of immunoglobulin light chains in tissue 456 Painful neuropathy Autonomic involvement Carpal tunnel syndrome Muscle amyloid Amyloidoma (trigeminal root) Secondary or reactive amyloidosis (AA) Chronic inflammatory diseases, rheumatoid diseases, osteomyelitis Deposition of acute phase plasma protein, serum amyloid A: polyneuropathy not -
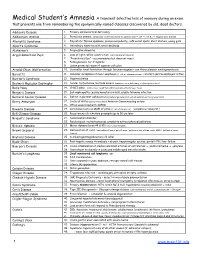
1 Medical Student's Amnesia a Transient Selective Loss of Memory During an Exam That Prevents One from Remem
Medical Student’s Amnesia A transient selective loss of memory during an exam that prevents one from remembering the eponymically-named diseases discovered by old, dead doctors. Addison’s Disease 1. Primary adrenocortical deficiency Addisonian Anemia 2. Pernicious anemia (antibodies to intrinsic factor or parietal cells → ↓IF → ↓Vit B12 → megaloblastic anemia) Albright’s Syndrome 3. Polyostotic fibrous dysplasia, precocious puberty, café au lait spots, short stature, young girls Alport’s Syndrome 4. Hereditary nephritis with nerve deafness Alzheimer’s 5. Progressive dementia Argyll-Robertson Pupil 6. Loss of light reflex constriction (contralateral or bilateral) 7. “Prostitute’s Eye” – accommodates but does not react 8. Pathognomonic for 3°Syphilis 9. Lesion pretectal region of superior colliculus Arnold-Chiari Malformation 10. Cerebellar tonsil herniation through foramen magnum = see thoracolumbar meningomyelocele Barrett’s 11. Columnar metaplasia of lower esophagus (↑ risk of adenocarcinoma)- constant gastroesophageal reflux Bartter’s Syndrome 12. Hyperreninemia Becker’s Muscular Dystrophy 13. Similar to Duchenne, but less severe (mutation, not a deficiency, in dystrophin protein) Bell’s Palsy 14. CNVII palsy (entire face; recall that UMN lesion only affects lower face) Berger’s Disease 15. IgA nephropathy causing hematuria in kids, usually following infection Bernard-Soulier Disease 16. Defect in platelet adhesion (abnormally large platelets & lack of platelet-surface glycoprotein) Berry Aneurysm 17. Circle of Willis (subarachnoid bleed) Anterior Communicating artery 18. Often associated with ADPKD Bowen’s Disease 19. Carcinoma in situ on shaft of penis (↑ risk of visceral ca) [compare w/ Queyrat] Brill-Zinsser Disease 20. Recurrences of rickettsia prowazaki up to 50 yrs later Briquet’s Syndrome 21. -

432 Medicine Team Leaders 1St Questions: D Raghad Al Mutlaq & Abdulrahman Al Zahrani 2Nd Questions: D for Mistakes Or Feedback: [email protected]
MEDICINE 432 Team 51 Myopathies Done By: Reviewed By: Abdurahman Alakeel Sarah Al-Essa COLOR GUIDE: • Females' Notes • Males' Notes • Important • Additional 432MedicineTeam Myopathies Objectives Not Given 1 432MedicineTeam Myopathies Myopathies: • Myo -is muscle, pathos is suffering in Greek • Disorders in which there is a primary functional (like problems in ion channels) or structural (destruction of muscle tissue replaced by connective tissue or fat) impairment of skeletal muscle. • Helpful link: https://www.youtube.com/watch?v=8wa04qYsaps Approach: • THREE STEPS: 1. Distinguishing true muscle weakness from other symptoms. • Distinguishing type of pain or weakness is the first step in evaluating patients with muscle-related complaints. • SOB (shortness of breath), joint pain (asthenia), fatigue, poor exercise tolerance, or paresthesia, rather than a true muscle weakness. • Heart failure, arthritis, depression… Asthenia: 2. Distinguishing CNS from PNS lesions. (PNS lesions present bilaterally) Motor impairment do to pain or joint 3. Determining the etiology. dysfunction SYMPTOMS: • Positive symptoms: Myalgia ,myotonia ,cramps ,contractures ,myoglobinuria. • Negative symptoms: Weakness, atrophy, exercise intolerance, periodic paralysis. 2 432MedicineTeam Myopathies 1- Weakness • Weakness is the cardinal symptom • The distribution of weakness is variable and may change over time . • Most of the time in the proximal muscles; muscles of the shoulder girdle. • Patients complain from difficulty arising from a chair or low toilet, difficulty climbing stairs, a waddling gait, difficulty lifting objects over the head, combing hair or brushing teeth. • Distal weakness is less common. • Patients with proximal leg weakness may rise from sitting on the floor by “climbing up their legs with their hands”, This is termed Gower's Gower’s sign sign.