Drug Interaction Studies on New Drug Applications: Current Situations and Regulatory Views in Japan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
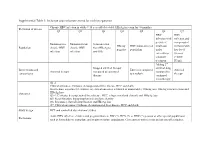
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection
pharmaceuticals Review Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection Isabella Zanella 1,2,* , Daniela Zizioli 1, Francesco Castelli 3 and Eugenia Quiros-Roldan 3 1 Department of Molecular and Translational Medicine, University of Brescia, 25123 Brescia, Italy; [email protected] 2 Clinical Chemistry Laboratory, Cytogenetics and Molecular Genetics Section, Diagnostic Department, ASST Spedali Civili di Brescia, Piazzale Spedali Civili 1, 25123 Brescia, Italy 3 University Department of Infectious and Tropical Diseases, University of Brescia and ASST Spedali Civili di Brescia, Piazzale Spedali Civili 1, 25123 Brescia, Italy; [email protected] (F.C.); [email protected] (E.Q.-R.) * Correspondence: [email protected]; Tel.: +39-030-399-6806 Abstract: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is spreading worldwide with different clinical manifestations. Age and comorbidities may explain severity in critical cases and people living with human immunodeficiency virus (HIV) might be at particularly high risk for severe progression. Nonetheless, current data, although sometimes contradictory, do not confirm higher morbidity, risk of more severe COVID-19 or higher mortality in HIV-infected people with complete access to antiretroviral therapy (ART). A possible protective role of ART has been hypothesized to explain these observations. Anti-viral drugs used to treat HIV infection have been repurposed for COVID-19 treatment; this is also based on previous studies on severe acute respiratory syndrome virus (SARS-CoV) and Middle East respiratory syndrome virus (MERS-CoV). Among Citation: Zanella, I.; Zizioli, D.; them, lopinavir/ritonavir, an inhibitor of viral protease, was extensively used early in the pandemic Castelli, F.; Quiros-Roldan, E. -

The Evolution of Antiviral Therapy for External Ocular Viral Infections Over Twenty-Five Years
Cornea 19(5): 673–680, 2000. © 2000 Lippincott Williams & Wilkins, Inc., Philadelphia The Evolution of Antiviral Therapy for External Ocular Viral Infections Over Twenty-five Years Y. Jerold Gordon, M.D. Purpose. To review the past 25 years of the evolution of antiviral levels on the corneal surface. By 1975, IDU was the treatment of therapy for the treatment of common external ocular viral infec- choice for herpetic epithelial keratitis worldwide. However, the tions (herpes simplex virus type 1, varicella-zoster virus, and ad- shortcomings of topical IDU therapy: significant toxicity (super- enovirus). Methods. A broad-based literature review in the fields ficial punctate keratitis, chemical conjunctivitis, and punctal oc- of virology, antiviral research, and ophthalmology will be carried clusion) and frequent hypersensitivity reactions, limited its useful- out. The pathogenesis of the major external ocular viral infections ness. As an insoluble agent, it had no therapeutic effect on herpetic and history of antiviral development will be cited. Important con- ceptual breakthroughs as well as historical landmarks will be em- stromal keratitis or iritis. Today, IDU is used infrequently in de- phasized. Results. The successful development of effective anti- veloped nations because it has been replaced by better drugs. How- virals to treat the most common external ocular viral infections ever, it remains effective and is used in developing nations where have dramatically reduced morbidity and sight loss. The immune cost issues are critical. pathogenesis of herpetic stromal keratitis is better understood. In the mid-1970s, several promising strategies to treat HSV Conclusions. Remarkable progress in the development of antiviral epithelial keratitis failed. -

Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2
viruses Review Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2 Lauren A. Sadowski 1,†, Rista Upadhyay 1,2,†, Zachary W. Greeley 1,‡ and Barry J. Margulies 1,3,* 1 Towson University Herpes Virus Lab, Department of Biological Sciences, Towson University, Towson, MD 21252, USA; [email protected] (L.A.S.); [email protected] (R.U.); [email protected] (Z.W.G.) 2 Towson University Department of Chemistry, Towson, MD 21252, USA 3 Molecular Biology, Biochemistry, and Bioinformatics Program, Towson University, Towson, MD 21252, USA * Correspondence: [email protected] † Authors contributed equally to this manuscript. ‡ Current address: Becton-Dickinson, Sparks, MD 21152, USA. Abstract: Herpes simplex viruses-1 and -2 (HSV-1 and -2) are two of the three human alphaher- pesviruses that cause infections worldwide. Since both viruses can be acquired in the absence of visible signs and symptoms, yet still result in lifelong infection, it is imperative that we provide interventions to keep them at bay, especially in immunocompromised patients. While numerous experimental vaccines are under consideration, current intervention consists solely of antiviral chemotherapeutic agents. This review explores all of the clinically approved drugs used to prevent the worst sequelae of recurrent outbreaks by these viruses. Keywords: acyclovir; ganciclovir; cidofovir; vidarabine; foscarnet; amenamevir; docosanol; nelfi- navir; HSV-1; HSV-2 Citation: Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections 1. Introduction with Herpes Simplex Viruses-1 and -2. The world of anti-herpes simplex (anti-HSV) agents took flight in 1962 with the FDA Viruses 2021, 13, 1228. -

Ep001156797b1*
(19) *EP001156797B1* (11) EP 1 156 797 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61P 31/22 (2006.01) A61P 31/20 (2006.01) 24.10.2007 Bulletin 2007/43 A61P 31/16 (2006.01) A61P 31/14 (2006.01) A61P 31/12 (2006.01) A61P 35/00 (2006.01) (2006.01) (2006.01) (21) Application number: 00912195.5 A61K 45/06 A61K 31/70 A61K 31/522 (2006.01) A61K 31/045 (2006.01) (22) Date of filing: 08.03.2000 (86) International application number: PCT/US2000/005965 (87) International publication number: WO 2000/053167 (14.09.2000 Gazette 2000/37) (54) SYNERGISTIC INHIBITION OF VIRAL REPLICATION BY LONG-CHAIN HYDROCARBONS AND NUCLEOSIDE ANALOGS SYNERGISTISCHE HEMMUNG VON VIRALREPLIKATION DURCH LANGKETTIGEN KOHLENWASSERSTOFFE UND NUCLEOSID-ANALOGE INHIBITION SYNERGIQUE DE REPLICATION VIRALE PAR DES HYDROCARBURES ET ANALOGUES NUCLEOSIDIQUES A CHAINE LONGUE (84) Designated Contracting States: • KATZ, David, H. AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU La Jolla, CA 92037 (US) MC NL PT SE • KATZ, Lee, R. Designated Extension States: La Jolla, CA 92037 (US) AL LT LV MK RO SI (74) Representative: W.P. Thompson & Co. (30) Priority: 10.03.1999 US 265922 Coopers Building Church Street (43) Date of publication of application: Liverpool L1 3AB (GB) 28.11.2001 Bulletin 2001/48 (56) References cited: (73) Proprietor: AVANIR PHARMACEUTICALS WO-A-95/16434 WO-A-96/40144 San Diego, California 92121 (US) WO-A-97/13528 WO-A-98/11887 WO-A-98/30244 US-A- 5 484 911 (72) Inventors: • MARCELLETTI, John, F. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

WHO Pharmaceuticals Newsletter Provides You
2018 WHO Pharmaceuticals No.6 NEWSLETTER The WHO Pharmaceuticals Newsletter provides you with the latest information on the safety of medicines WHO Vision for Medicines Safety and legal actions taken by regulatory authorities around No country left behind: the world. It also provides signals based on information worldwide pharmacovigilance for safer medicines, safer patients derived from the WHO global database of individual case safety reports, VigiBase. This newsletter also includes a short report from the The aim of the Newsletter is to disseminate regulatory 41st Annual Meeting of Representatives of National information on the safety of Pharmacovigilance Centres participating in the WHO pharmaceutical products, Programme for International Drug Monitoring and from based on communications received from our network of an Advanced Workshop for Strengthening national pharmacovigilance centres Pharmacovigilance (PV) Systems. and other sources such as specialized bulletins and journals, as well as partners in WHO. The information is produced in the form of résumés in English, full texts of which may be obtained on request from: Safety and Vigilance: Medicines, EMP-HIS, World Health Organization, 1211 Geneva 27, Switzerland, Contents E-mail address: [email protected] This Newsletter is also available at: Regulatory matters http://www.who.int/medicines Safety of medicines Signal Feature © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. -

Chronic Varicella-Zoster Virus Epithelial Keratitis in Patients with Acquired Immunodeficiency Syndrome
CLINICAL SCIENCES Chronic Varicella-zoster Virus Epithelial Keratitis in Patients With Acquired Immunodeficiency Syndrome Kenneth C. Chern, MD; Diana Conrad, MBBS, FRACO; Gary N. Holland, MD; Douglas S. Holsclaw, MD; Lee K. Schwartz, MD; Todd P. Margolis, MD, PhD Objective: To characterize further a chronic epithelial fluorescein and rose bengal. The peripheral and midpe- keratitis caused by varicella-zoster virus infection in pa- ripheral cornea was most commonly affected, and, in 13 tients with acquired immunodeficiency syndrome (AIDS). of the 16 patients, the lesions crossed the limbus. Pain was a prominent feature, occurring in 12 of 16 patients. Methods: Patients with AIDS and chronic epithelial In 9 of 12 patients tested, varicella-zoster virus was iden- keratitis associated with varicella-zoster virus from 3 in- tified by culture, direct fluorescent antibody testing, poly- stitutions were identified. Patient records were reviewed merase chain reaction testing, or a combination of these retrospectively for the following data: medical and demo- studies, with direct fluorescent antibody testing (6 of 8 graphic characteristics, techniques of diagnosis, physical positive results) and polymerase chain reaction testing findings, course, response to treatment, and outcome. (3 of 3 positive results) appearing to be the most sensi- tive. Response to antiviral medication was variable. Results: Sixteen patients were studied. CD4+ T- lymphocyte cell counts were available in 11 patients, with Conclusions: In patients with AIDS, varicella-zoster vi- a median of 0.034 3 109/L (range, 0-0.094 3 109/L ). Two rus may cause a chronic infection of the corneal epithe- patients had no history of a zosteriform rash. -

Advances and Perspectives in the Management of Varicella-Zoster Virus Infections
molecules Review Advances and Perspectives in the Management of Varicella-Zoster Virus Infections Graciela Andrei * and Robert Snoeck Laboratory of Virology and Chemotherapy, Rega Institute for Medical Research, KU Leuven, 3000 Leuven, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32-16-32-19-51 Abstract: Varicella-zoster virus (VZV), a common and ubiquitous human-restricted pathogen, causes a primary infection (varicella or chickenpox) followed by establishment of latency in sensory ganglia. The virus can reactivate, causing herpes zoster (HZ, shingles) and leading to significant morbidity but rarely mortality, although in immunocompromised hosts, VZV can cause severe disseminated and occasionally fatal disease. We discuss VZV diseases and the decrease in their incidence due to the introduction of live-attenuated vaccines to prevent varicella or HZ. We also focus on acyclovir, valacyclovir, and famciclovir (FDA approved drugs to treat VZV infections), brivudine (used in some European countries) and amenamevir (a helicase-primase inhibitor, approved in Japan) that augur the beginning of a new era of anti-VZV therapy. Valnivudine hydrochloride (FV-100) and valomaciclovir stearate (in advanced stage of development) and several new molecules potentially good as anti-VZV candidates described during the last year are examined. We reflect on the role of antiviral agents in the treatment of VZV-associated diseases, as a large percentage of the at-risk population is not immunized, and on the limitations of currently FDA-approved anti-VZV drugs. Their low efficacy in controlling HZ pain and post-herpetic neuralgia development, and the need of Citation: Andrei, G.; Snoeck, R. multiple dosing regimens requiring daily dose adaptation for patients with renal failure urges the Advances and Perspectives in the development of novel anti-VZV drugs. -

Repurposing of FDA Approved Drugs
Antiviral Drugs (In Phase IV) ABACAVIR GEMCITABINE ABACAVIR SULFATE GEMCITABINE HYDROCHLORIDE ACYCLOVIR GLECAPREVIR ACYCLOVIR SODIUM GRAZOPREVIR ADEFOVIR DIPIVOXIL IDOXURIDINE AMANTADINE IMIQUIMOD AMANTADINE HYDROCHLORIDE INDINAVIR AMPRENAVIR INDINAVIR SULFATE ATAZANAVIR LAMIVUDINE ATAZANAVIR SULFATE LEDIPASVIR BALOXAVIR MARBOXIL LETERMOVIR BICTEGRAVIR LOPINAVIR BICTEGRAVIR SODIUM MARAVIROC BOCEPREVIR MEMANTINE CAPECITABINE MEMANTINE HYDROCHLORIDE CARBARIL NELFINAVIR CIDOFOVIR NELFINAVIR MESYLATE CYTARABINE NEVIRAPINE DACLATASVIR OMBITASVIR DACLATASVIR DIHYDROCHLORIDE OSELTAMIVIR DARUNAVIR OSELTAMIVIR PHOSPHATE DARUNAVIR ETHANOLATE PARITAPREVIR DASABUVIR PENCICLOVIR DASABUVIR SODIUM PERAMIVIR DECITABINE PERAMIVIR DELAVIRDINE PIBRENTASVIR DELAVIRDINE MESYLATE PODOFILOX DIDANOSINE RALTEGRAVIR DOCOSANOL RALTEGRAVIR POTASSIUM DOLUTEGRAVIR RIBAVIRIN DOLUTEGRAVIR SODIUM RILPIVIRINE DORAVIRINE RILPIVIRINE HYDROCHLORIDE EFAVIRENZ RIMANTADINE ELBASVIR RIMANTADINE HYDROCHLORIDE ELVITEGRAVIR RITONAVIR EMTRICITABINE SAQUINAVIR ENTECAVIR SAQUINAVIR MESYLATE ETRAVIRINE SIMEPREVIR FAMCICLOVIR SIMEPREVIR SODIUM FLOXURIDINE SOFOSBUVIR FOSAMPRENAVIR SORIVUDINE FOSAMPRENAVIR CALCIUM STAVUDINE FOSCARNET TECOVIRIMAT FOSCARNET SODIUM TELBIVUDINE GANCICLOVIR TENOFOVIR ALAFENAMIDE GANCICLOVIR SODIUM TENOFOVIR ALAFENAMIDE FUMARATE TIPRANAVIR VELPATASVIR TRIFLURIDINE VIDARABINE VALACYCLOVIR VOXILAPREVIR VALACYCLOVIR HYDROCHLORIDE ZALCITABINE VALGANCICLOVIR ZANAMIVIR VALGANCICLOVIR HYDROCHLORIDE ZIDOVUDINE Antiviral Drugs (In Phase III) ADEFOVIR LANINAMIVIR OCTANOATE -

WO 2013/167743 Al 14 November 2013 (14.11.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2013/167743 Al 14 November 2013 (14.11.2013) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/18 (2006.01) A61K 31/708 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A61K 31/522 (2006.01) A61K 45/06 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 31/675 (2006.01) A61P 29/00 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, A61K 31/7068 (2006.01) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (21) International Application Number: NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, PCT/EP2013/059752 RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, (22) International Filing Date: TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, 10 May 2013 (10.05.2013) ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 12167771 .0 11 May 2012 ( 11.05.2012) EP TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, (71) Applicant: AKRON MOLECULES GMBH [AT/AT]; MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, Helmut-Qualtinger-Gasse 2, A-1030 Vienna (AT). -
Patient Information Leaflet
PACKAGE LEAFLET: INFORMATION FOR THE USER Fluorouracil 25 mg/ml Injection Read all of this leaflet carefully before you start taking this medicine. • Keep this leaflet. You may need to read it again. • If you have any further questions, ask your doctor or pharmacist. • This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their symptoms are the same as yours. • If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet: 1. What Fluorouracil Injection is and what it is used for 2. What you need to know before you use Fluorouracil Injection 3. How to use Fluorouracil Injection 4. Possible side effects 5. How to store Fluorouracil Injection 6. Contents of the pack and other information 1. What Fluorouracil Injection is and what it is used for Fluorouracil Injection is an anti-cancer medicine. Treatment with an anti-cancer medicine is sometimes called cancer chemotherapy. Fluorouracil Injection is used to treat many common cancers, particularly cancers of the large bowel and breast. It may be used in combination with other anti-cancer medicines or radiotherapy. 2. What you need to know before you use Fluorouracil Injection Do not use Fluorouracil Injection • if you are allergic to Fluorouracil or any of the other ingredients of this medicine (listed in section 6) • if you are in a seriously weakened (including nutritional) state due to long illness • if your bone marrow has been damaged by other cancer treatments (including radiotherapy) • if you know that you do not have any activity of the enzyme dihydropyrimidine dehydrogenase (DPD) (complete DPD deficiency) • if you have a potentially serious infection • if your cancer is non-malignant • if you are pregnant or you are breast-feeding • if you are taking or have taken in the past 4 weeks brivudine, sorivudine and similar drugs (antivirals) Tell your doctor if any of the above applies to you before this medicine is used.