14-17 January 2019 PRAC Meeting Minutes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Protean Neurologic Manifestations of Varicella-Zoster Virus Infection
REVIEW MARIA A. NAGEL, MD DONALD H. GILDEN, MD Department of Neurology, University of Departments of Neurology and Microbiology, Colorado Health Sciences Center, Denver University of Colorado Health Sciences Center, Denver The protean neurologic manifestations of varicella-zoster virus infection ■ ABSTRACT ARICELLA-ZOSTER VIRUS (VZV) is an V exclusively human, highly neurotropic Multiple neurologic complications may follow the alphaherpesvirus. Primary infection causes reactivation of varicella-zoster virus (VZV), including chickenpox, after which the virus becomes herpes zoster (also known as zoster or shingles), latent in cranial nerve ganglia, dorsal root postherpetic neuralgia, vasculopathy, myelitis, necrotizing ganglia, and autonomic ganglia along the retinitis, and zoster sine herpete (pain without rash). entire neuraxis. Decades later, VZV can reac- These conditions can be difficult to recognize, especially tivate to cause a number of neurologic condi- as several can occur without rash. tions. This article discusses the protean manifes- ■ KEY POINTS tations of VZV reactivation and their diagno- sis and treatment. The most common sequela of herpes zoster is postherpetic neuralgia, which can persist for months and ■ VZV REACTIVATION sometimes years after the rash resolves. HAS MANY MANIFESTATIONS VZV vasculopathy can present as transient ischemic VZV reactivation can produce a number of neurologic complications: attacks, ischemic or hemorrhagic stroke, or aneurysm. •Herpes zoster (shingles), manifesting as pain and rash in up to three dermatomes Vasculopathy and neurologic complications of VZV are • Postherpetic neuralgia, the most common best diagnosed by detecting VZV DNA in bodily fluids or sequela of zoster, is pain that persists for tissues or anti-VZV immunoglobulin G in the months and sometimes years after the rash cerebrospinal fluid. -
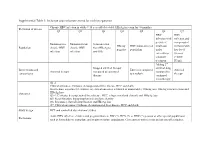
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions
Review CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions Rossana Roncato 1,*,†, Jacopo Angelini 2,†, Arianna Pani 2,3,†, Erika Cecchin 1, Andrea Sartore-Bianchi 2,4, Salvatore Siena 2,4, Elena De Mattia 1, Francesco Scaglione 2,3,‡ and Giuseppe Toffoli 1,‡ 1 1 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico (CRO), IRCCS, 33081 Aviano, Italy; [email protected] (E.C.); [email protected] (E.D.M.); [email protected] (G.T.) 2 Department of Oncology and Hemato-Oncology, Università degli Studi di Milano, 20122 Milan, Italy; [email protected] (J.A.); [email protected] (A.P.); [email protected] (A.S-B.); [email protected] (S.S.); [email protected] (F.S.) 3 Clinical Pharmacology Unit, ASST Grande Ospedale Metropolitano Niguarda, Piazza dell'Ospedale Maggiore 3, 20162 Milan, Italy 4 Department of Hematology and Oncology, Niguarda Cancer Center, Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy * Correspondence: [email protected]; Tel.:+390434659130 † These authors contributed equally. ‡ These authors share senior authorship. Int. J. Mol. Sci. 2020, 21, x; doi: www.mdpi.com/journal/ijms Int. J. Mol. Sci. 2020, 21, x 2 of 8 Table S1. Co-administered agents categorized according to their potential risk for Drug-Drug interaction (DDI) in combination with CDK4/6 inhibitors (CDKis). Colors suggest the risk of DDI with CDKis: green, low risk DDI; orange, moderate risk DDI; red, high risk DDI. ADME, absorption, distribution, metabolism, and excretion; GI, Gastrointestinal; TdP, Torsades de Pointes; NTI, narrow therapeutic index. * Cardiological toxicity should be considered especially for ribociclib due to the QT prolongation. -

Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy Using Machine Learning
Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy using Machine Learning Kate Wang et al. Supplemental Table S1. Drugs selected by Pharmacophore-based, ML-based and DL- based search in the FDA-approved drugs database Pharmacophore WEKA TF 1-Palmitoyl-2-oleoyl-sn-glycero-3- 5-O-phosphono-alpha-D- (phospho-rac-(1-glycerol)) ribofuranosyl diphosphate Acarbose Amikacin Acetylcarnitine Acetarsol Arbutamine Acetylcholine Adenosine Aldehydo-N-Acetyl-D- Benserazide Acyclovir Glucosamine Bisoprolol Adefovir dipivoxil Alendronic acid Brivudine Alfentanil Alginic acid Cefamandole Alitretinoin alpha-Arbutin Cefdinir Azithromycin Amikacin Cefixime Balsalazide Amiloride Cefonicid Bethanechol Arbutin Ceforanide Bicalutamide Ascorbic acid calcium salt Cefotetan Calcium glubionate Auranofin Ceftibuten Cangrelor Azacitidine Ceftolozane Capecitabine Benserazide Cerivastatin Carbamoylcholine Besifloxacin Chlortetracycline Carisoprodol beta-L-fructofuranose Cilastatin Chlorobutanol Bictegravir Citicoline Cidofovir Bismuth subgallate Cladribine Clodronic acid Bleomycin Clarithromycin Colistimethate Bortezomib Clindamycin Cyclandelate Bromotheophylline Clofarabine Dexpanthenol Calcium threonate Cromoglicic acid Edoxudine Capecitabine Demeclocycline Elbasvir Capreomycin Diaminopropanol tetraacetic acid Erdosteine Carbidopa Diazolidinylurea Ethchlorvynol Carbocisteine Dibekacin Ethinamate Carboplatin Dinoprostone Famotidine Cefotetan Dipyridamole Fidaxomicin Chlormerodrin Doripenem Flavin adenine dinucleotide -

New Zealand Data Sheet – Tradename (Active Ingredient)
NEW ZEALAND DATA SHEET NEW ZEALAND DATA SHEET FLUOROURACIL EBEWE (FLUOROURACIL) 1. PRODUCT NAME Fluorouracil Ebewe 50 mg/mL solution for injection 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Fluorouracil Ebewe 250 mg/5 mL contains 250 mg fluorouracil*. Fluorouracil Ebewe 500 mg/10 mL contains 100 mg fluorouracil*. Fluorouracil Ebewe 1000 mg/20 mL contains 1000 mg fluorouracil. Fluorouracil Ebewe 2500 mg/50 mL contains 2500 mg fluorouracil. Fluorouracil Ebewe 5000 mg/100 mL contains 5000 mg fluorouracil. For the full list of excipients, see Section 6.1 List of excipients. *Not supplied in New Zealand 3. PHARMACEUTICAL FORM Fluorouracil Ebewe is a sterile solution of fluorouracil for use as an intravenous infusion or injection. The pH of the fluorouracil injection solution is approximately 8.9. 4. CLINICAL PARTICULARS 4.1. THERAPEUTIC INDICATIONS Alone or in combination, for the palliative treatment of malignant tumours, particularly of the breast, colon or rectum; and in the treatment of gastric, primary hepatic, pancreatic, uterine (cervical particularly), ovarian and bladder carcinomas. Fluorouracil should only be used when other proven measures have failed or are considered impractical. 4.2. DOSE AND METHOD OF ADMINISTRATION General Directions Fluorouracil Ebewe contains no antimicrobial agent. The product is for single use in one patient only. Discard any residue. To reduce microbiological hazard, use as soon as practicable after reconstitution/preparation. If storage is necessary, hold at 2°C to 8°C for not more than 24 hours after preparation. Administration should be completed within 24 hours of preparation of the infusion and any residue discarded according to the guidelines for the disposal of cytotoxic drugs (see Section 6.6 Special precautions for disposal). -

Clinical Practice Guidelines for Irritable Bowel Syndrome in Korea, 2017 Revised Edition
J Neurogastroenterol Motil, Vol. 24 No. 2 April, 2018 pISSN: 2093-0879 eISSN: 2093-0887 https://doi.org/10.5056/jnm17145 JNM Journal of Neurogastroenterology and Motility Review Clinical Practice Guidelines for Irritable Bowel Syndrome in Korea, 2017 Revised Edition Kyung Ho Song,1,2 Hye-Kyung Jung,3* Hyun Jin Kim,4 Hoon Sup Koo,1 Yong Hwan Kwon,5 Hyun Duk Shin,6 Hyun Chul Lim,7 Jeong Eun Shin,6 Sung Eun Kim,8 Dae Hyeon Cho,9 Jeong Hwan Kim,10 Hyun Jung Kim11; and The Clinical Practice Guidelines Group Under the Korean Society of Neurogastroenterology and Motility 1Department of Internal Medicine, Konyang University College of Medicine, Daejeon, Korea; 2Konyang University Myunggok Medical Research Institute Daejeon, Korea; 3Department of Internal Medicine, Ewha Womans University School of Medicine, Seoul, Korea; 4Department of Internal Medicine, Gyeongsang National University, College of Medicine, Jinju, Korea; 5Department of Internal Medicine, Kyungpook National University, School of Medicine, Daegu, Korea; 6Department of Internal Medicine, Dankook University College of Medicine, Cheonan, Korea; 7Department of Internal Medicine, Yongin Severance Hospital, Yonsei University College of Medicine, Yongin, Korea; 8Department of Internal Medicine, Kosin University College of Medicine, Busan, Korea; 9Department of Internal Medicine, Sungkyunkwan University School of Medicine, Changwon, Korea; 10Department of Internal Medicine, Konkuk University School of Medicine, Seoul, Korea; and 11Department of Preventive Medicine, Korea University College of Medicine, Seoul, Korea In 2011, the Korean Society of Neurogastroenterology and Motility (KSNM) published clinical practice guidelines on the management of irritable bowel syndrome (IBS) based on a systematic review of the literature. The KSNM planned to update the clinical practice guidelines to support primary physicians, reduce the socioeconomic burden of IBS, and reflect advances in the pathophysiology and management of IBS. -

1: Gastro-Intestinal System
1 1: GASTRO-INTESTINAL SYSTEM Antacids .......................................................... 1 Stimulant laxatives ...................................46 Compound alginate products .................. 3 Docuate sodium .......................................49 Simeticone ................................................... 4 Lactulose ....................................................50 Antimuscarinics .......................................... 5 Macrogols (polyethylene glycols) ..........51 Glycopyrronium .......................................13 Magnesium salts ........................................53 Hyoscine butylbromide ...........................16 Rectal products for constipation ..........55 Hyoscine hydrobromide .........................19 Products for haemorrhoids .................56 Propantheline ............................................21 Pancreatin ...................................................58 Orphenadrine ...........................................23 Prokinetics ..................................................24 Quick Clinical Guides: H2-receptor antagonists .......................27 Death rattle (noisy rattling breathing) 12 Proton pump inhibitors ........................30 Opioid-induced constipation .................42 Loperamide ................................................35 Bowel management in paraplegia Laxatives ......................................................38 and tetraplegia .....................................44 Ispaghula (Psyllium husk) ........................45 ANTACIDS Indications: -

PRODUCT INFORMATION Brivudine Item No
PRODUCT INFORMATION Brivudine Item No. 29448 CAS Registry No.: 69304-47-8 Formal Name: 5-[(1E)-2-bromoethenyl]-2′-deoxy-uridine OH Synonym: BVDU OH MF: C11H13BrN2O5 H Br FW: 333.1 N O Purity: ≥98% UV/Vis.: λ: 251, 294 nm max OO Supplied as: A crystalline solid N Storage: -20°C H Stability: ≥2 years Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Brivudine is supplied as a crystalline solid. A stock solution may be made by dissolving the brivudine in the solvent of choice, which should be purged with an inert gas. Brivudine is soluble in organic solvents such as ethanol, DMSO, and dimethyl formamide (DMF). The solubility of brivudine in ethanol is approximately 10 mg/ml and approximately 30 mg/ml in DMSO and DMF. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant, since organic solvents may have physiological effects at low concentrations. Organic solvent-free aqueous solutions of brivudine can be prepared by directly dissolving the crystalline solid in aqueous buffers. The solubility of brivudine in PBS, pH 7.2, is approximately 0.5 mg/ml. We do not recommend storing the aqueous solution for more than one day. Description Brivudine is a thymidine analog and inhibitor of herpes simplex virus 1 (HSV-1) and varicella-zoster virus (VZV) viral replication.1,2 Following conversion to a 5’-triphosphate form by viral kinases, brivudine is incorporated into viral DNA, inhibiting viral DNA polymerases and inducing strand breakage.2 Brivudine (0.02-0.04 μg/ml) inhibits VZV replication in primary human fibroblasts and inhibits cytopathogenic 1,3 effects of the KOS HSV-1 strain in a panel of cell lines (MIC50s = 0.007-0.4 μg/ml). -

Efficacy of the Combination of Pinaverium Bromide 100Mg Plus Simethicone 300Mg in Abdominal Pain and Bloating in Irritable Bowel
University of Texas Rio Grande Valley ScholarWorks @ UTRGV School of Medicine Publications and Presentations School of Medicine 4-2020 Efficacy of the Combination of Pinaverium Bromide 100mg Plus Simethicone 300mg in Abdominal Pain and Bloating in Irritable Bowel Syndrome: A Randomized, Placebo-controlled Trial Max J. Schmulson Jazmin Chiu-Ugalde Adolfo Saez-Rios Aurelio Lopez-Colombo Gualberto J. Mateos-Perez See next page for additional authors Follow this and additional works at: https://scholarworks.utrgv.edu/som_pub Part of the Chemicals and Drugs Commons Recommended Citation Schmulson, M. J., Chiu-Ugalde, J., Sáez-Ríos, A., López-Colombo, A., Mateos-Pérez, G. J., Remes-Troche, J. M., Sobrino-Cossio, S., Soto-Pérez, J. C., Tamayo de la Cuesta, J. L., Teramoto-Matsubara, O. T., & López-Alvarenga, J. C. (2020). Efficacy of the Combination of Pinaverium Bromide 100 mg Plus Simethicone 300 mg in Abdominal Pain and Bloating in Irritable Bowel Syndrome: A Randomized, Placebo-controlled Trial. Journal of Clinical Gastroenterology, 54(4), e30–e39. https://doi.org/10.1097/ MCG.0000000000001242 This Article is brought to you for free and open access by the School of Medicine at ScholarWorks @ UTRGV. It has been accepted for inclusion in School of Medicine Publications and Presentations by an authorized administrator of ScholarWorks @ UTRGV. For more information, please contact [email protected], [email protected]. Authors Max J. Schmulson, Jazmin Chiu-Ugalde, Adolfo Saez-Rios, Aurelio Lopez-Colombo, Gualberto J. Mateos- Perez, Jose Mario Remes-Troche, Sergio Sobrino-Cossio, Julio C. Soto-Perez, Jose L. Tamayo de la Cuesta, Oscar T. Teramoto-Matsubara, and Juan C. -

Silibinin Component for the Treatment of Hepatitis Silbininkomponente Zur Behandlung Von Hepatitis Composant De Silibinine Pour Le Traitement De L’Hépatite
(19) & (11) EP 2 219 642 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 31/357 (2006.01) A61P 1/16 (2006.01) 21.09.2011 Bulletin 2011/38 A61P 31/12 (2006.01) (21) Application number: 08849759.9 (86) International application number: PCT/EP2008/009659 (22) Date of filing: 14.11.2008 (87) International publication number: WO 2009/062737 (22.05.2009 Gazette 2009/21) (54) SILIBININ COMPONENT FOR THE TREATMENT OF HEPATITIS SILBININKOMPONENTE ZUR BEHANDLUNG VON HEPATITIS COMPOSANT DE SILIBININE POUR LE TRAITEMENT DE L’HÉPATITE (84) Designated Contracting States: (56) References cited: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR WO-A-02/067853 GB-A- 2 167 414 HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR • POLYAK STEPHEN J ET AL: "Inhibition of T- cell inflammatory cytokines, hepatocyte NF-kappa B (30) Priority: 15.11.2007 EP 07022187 signaling, and HCV infection by standardized 15.11.2007 US 988168 P silymarin" GASTROENTEROLOGY, vol. 132, no. 25.03.2008 EP 08005459 5, May 2007 (2007-05), pages 1925-1936, XP002477920 ISSN: 0016-5085 (43) Date of publication of application: • MAYER K E ET AL: "Silymarin treatment of viral 25.08.2010 Bulletin 2010/34 hepatitis: A systematic review" JOURNAL OF VIRAL HEPATITIS 200511 GB, vol. 12, no. 6, (60) Divisional application: November 2005 (2005-11), pages 559-567, 11005445.9 XP002477921 ISSN: 1352-0504 1365-2893 • CHAVEZ M L: "TREATMENT OF HEPATITIS C (73) Proprietor: Madaus GmbH WITH MILK THISTLE?" JOURNAL OF HERBAL 51067 Köln (DE) PHARMACOTHERAPY, HAWORTH HERBAL PRESS, BINGHAMTON, US, vol. -

HML Update, Effective 1 August 2018
Pharmaceutical Management Agency Section H Update for Hospital Pharmaceuticals Effective 1 August 2018 Contents Summary of decisions effective 1 August 2018 ............................................. 3 Section H changes to Part II .......................................................................... 8 Index ........................................................................................................... 16 2 Summary of decisions EFFECTIVE 1 AUGUST 2018 • Aluminium hydroxide with magnesium hydroxide and simeticone (e.g. Mylanta) tab 200 mg with magnesium hydroxide 200 mg and simeticone 20 mg – amended chemical name and presentation description) • Aluminium hydroxide with magnesium hydroxide and simeticone (e.g. Mylanta Double Strength) oral liq 400 mg with magnesium hydroxide 400 mg and simeticone 30 mg per 5 ml – amended chemical name and presentation description) • Aqueous cream (Pharmacy Health SLS-free) crm 100 g – price increase and addition of HSS • Atropine sulphate (Martindale) inj 600 mcg per ml, 1 ml ampoule – new listing and addition of HSS • Atropine sulphate (AstraZeneca) inj 600 mcg per ml, 1 ml ampoule – to be delisted 1 October 2018 • Baclofen (Pacifen) tab 10 mg – price increase and addition of HSS • Beractant (Survanta) soln 200 mg per 8 ml vial – to be delisted 1 January 2019 • Betamethasone valerate crm 0.1%, 50 g (Beta Cream) and oint 0.1%, 50 g (Beta Ointment) – price increase and addition of HSS • Betamethasone valerate (Beta Scalp) scalp app 0.1%, 100 ml – addition of HSS • Bosentan (Bosentan Dr -

Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection
pharmaceuticals Review Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection Isabella Zanella 1,2,* , Daniela Zizioli 1, Francesco Castelli 3 and Eugenia Quiros-Roldan 3 1 Department of Molecular and Translational Medicine, University of Brescia, 25123 Brescia, Italy; [email protected] 2 Clinical Chemistry Laboratory, Cytogenetics and Molecular Genetics Section, Diagnostic Department, ASST Spedali Civili di Brescia, Piazzale Spedali Civili 1, 25123 Brescia, Italy 3 University Department of Infectious and Tropical Diseases, University of Brescia and ASST Spedali Civili di Brescia, Piazzale Spedali Civili 1, 25123 Brescia, Italy; [email protected] (F.C.); [email protected] (E.Q.-R.) * Correspondence: [email protected]; Tel.: +39-030-399-6806 Abstract: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is spreading worldwide with different clinical manifestations. Age and comorbidities may explain severity in critical cases and people living with human immunodeficiency virus (HIV) might be at particularly high risk for severe progression. Nonetheless, current data, although sometimes contradictory, do not confirm higher morbidity, risk of more severe COVID-19 or higher mortality in HIV-infected people with complete access to antiretroviral therapy (ART). A possible protective role of ART has been hypothesized to explain these observations. Anti-viral drugs used to treat HIV infection have been repurposed for COVID-19 treatment; this is also based on previous studies on severe acute respiratory syndrome virus (SARS-CoV) and Middle East respiratory syndrome virus (MERS-CoV). Among Citation: Zanella, I.; Zizioli, D.; them, lopinavir/ritonavir, an inhibitor of viral protease, was extensively used early in the pandemic Castelli, F.; Quiros-Roldan, E.