Ep001156797b1*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interbiotech Entecavir
InterBioTech FT-XLS250 Entecavir Product Description Catalog #: XLS250, 5mg XLS251, 10mg XLS252, 50mg XLS253, 100mg AXAIV0, 1ml 10mM in DMSO. Catalog #: Name: Entecavir, Monohydrate Syn: BMS200475 monohydrate; SQ34676 monohydrate CAS : 209216-23-9 MW : 295.29 Formula : C12H17N5O4 Properties : DMSO : ≥ 50 mg/mL (169.33 mM) H2O : 2.8 mg/mL (9.48 mM) >99.5% Storage: Powder: -20°C (long term; possible at +4°C (2 years) (M) Also available: In solvent: -80°C (6 months) -20°C (1 month) Entecavir free form #RO893P/Q/R (Syn.: BMS200475; SQ34676) CAS No. : 142217-69-4; MW: 277.2 For Research Use Only Introduction Entecavir monohydrate (BMS200475 monohydrate; SQ34676 monohydrate) is a potent and selective inhibitor of HBV, with an EC50 of 3.75 nM in HepG2 cell. IC50 & Target EC50:3.75 nM (anti-HBV, HepG2 cell)[1] In Vitro *Solubility : DMSO : ≥ 50 mg/mL (169.33 mM) H2O : 2.8 mg/mL (9.48 mM; Need ultrasonic and warming) *Preparation : 1mM = 1mg in 3.3865 mL Entecavir monohydrate (BMS200475 monohydrate; SQ34676 monohydrate) has a EC50 of 3.75 nM against HBV. It is incorporated into the protein primer of HBV and subsequently inhibits the priming step of the reverse transcriptase. The antiviral activity of BMS-200475 is significantly less against the other RNA and DNA viruses[1]. Entecavir monohydrate is more readily phosphorylated to its active metabolites than other deoxyguanosine analogs (penciclovir, ganciclovir, lobucavir, and aciclovir) or lamivudine. The intracellular half-life of entecavir is 15 h[2]. P.1 InterBioTech FT-XLS250 In Vivo *Preparation : 1. Add each solvent one by one: 10% DMSO 40% PEG300 5% Tween-80 45% saline Solubility: ≥ 3 mg/mL (10.16 mM); Clear solution 2. -
Inclusion and Exclusion Criteria for Each Key Question
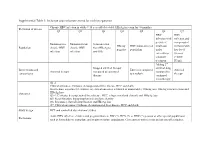
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Chemical Properties Biological Description Solubility Information
Data Sheet (Cat.No.T0085L) Entecavir Chemical Properties CAS No.: 142217-69-4 Formula: C12H15N5O3 Molecular Weight: 277.28 Appearance: Solid Storage: 0-4℃ for short term (days to weeks), or -20℃ for long term (months). Biological Description Description Entecavir is a guanosine nucleoside analogue used in the treatment of chronic hepatitis B virus (HBV) infection. Entecavir therapy can be associated with flares of the underlying hepatitis B during or after therapy, but has not been linked to cases of clinically apparent liver injury. Targets(IC50) HBV( HepG2 cell): EC50:3.75nM In vitro BMS-200475 has a EC50 of 3.75 nM against HBV. It is incorporated into the protein primer of HBV and subsequently inhibits the priming step of the reverse transcriptase. The antiviral activity of BMS-200475 is significantly less against the other RNA and DNA viruses[1]. Entecavir is more readily phosphorylated to its active metabolites than other deoxyguanosine analogs (penciclovir, ganciclovir, lobucavir, and aciclovir) or lamivudine. The intracellular half-life of entecavir is 15 h[2]. In vivo Daily oral treatment with BMS-200475 at doses ranging from 0.02 to 0.5 mg/kg of body weight for 1 to 3 months effectively reduces the level of woodchuck hepatitis virus (WHV) viremia in chronically infected woodchucks[3]. Cell Research BMS 200475 is prepared in phosphate-buffered saline (PBS) and diluted with appropriate medium containing 2% fetal bovine serum. HepG2 2.2.15 cells are plated at a density of 5×105 cells per well on 12-well Biocoat collagen-coated plates and are maintained in a confluent state for 2 to 3 days before being overlaid with 1 mL of medium spiked with BMS 200475. -

Direct Acting Antivirals for the Treatment of Chronic Viral Hepatitis
Hindawi Publishing Corporation Scienti�ca Volume 2012, Article ID 478631, 22 pages http://dx.doi.org/10.6064/2012/478631 Review Article Direct Acting Antivirals for the Treatment of Chronic Viral Hepatitis Peter Karayiannis Section of Hepatology and Gastroenterology, Department of Medicine, Imperial College, St Mary’s Campus, London W2 1PG, UK Correspondence should be addressed to Peter Karayiannis; [email protected] Received 17 September 2012; Accepted 8 October 2012 Academic Editors: M. Clementi and W. Vogel Copyright © 2012 Peter Karayiannis. is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. e development and evaluation of antiviral agents through carefully designed clinical trials over the last 25 years have heralded a new dawn in the treatment of patients chronically infected with the hepatitis B and C viruses, but not so for the D virus (HBV, HCV, and HDV). e introduction of direct acting antivirals (DDAs) for the treatment of HBV carriers has permitted the long- term use of these compounds for the continuous suppression of viral replication, whilst in the case of HCV in combination with the standard of care [SOC, pegylated interferon (PegIFN), and ribavirin] sustained virological responses (SVRs) have been achieved with increasing frequency. Progress in the case of HDV has been slow and lacking in signi�cant breakthroughs.is paper aims to summarise the current state of play in treatment approaches for chonic viral hepatitis patients and future perspectives. 1. Introduction recombinant subsequently, both of which have more recently been superceded by the pegylated form (PegIFN), which Conservative estimates of the number of individuals world- requires intramuscular injection only once a week as opposed wide who are thought to be chronically infected with either to three times a week with the previous forms. -
![Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Scaffold †](https://docslib.b-cdn.net/cover/4863/homocarbocyclic-nucleoside-analogs-with-an-optically-active-substituted-bicyclo-2-2-1-heptane-scaffold-654863.webp)
Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Scaffold †
Proceeding Paper 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Scaffold † Constantin I. Tănase 1,*, Constantin Drăghici 2, Anamaria Hanganu 2, Lucia Pintilie 1, Maria Maganu 2, Vladimir V. Zarubaev 3, Alexandrina Volobueva 3 and Ekaterina Sinegubova 3 1 National Institute for Chemical-Pharmaceutical Research and Development, 112 Vitan Av., 031299 Bucharest 3, Romania; [email protected] 2 Organic Chemistry Center “C.D.Nenitescu”, 202 B Splaiul Independentei, 060023 Bucharest, Romania; [email protected] (C.D.); [email protected] (A.H.); [email protected] (M.M.) 3 Department of Virology, Pasteur Institute of Epidemiology and Microbiology, 197101 St. Petersburg, Russia; [email protected] (V.V.Z.); [email protected] (A.V.); [email protected] (E.S.) * Correspondence: [email protected] † Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/. Abstract: An optically active bicyclo[2.2.0]heptane fragment was introduced in the molecule of new 1′-homonucleosides on a 2- 6-chloro-amino-purine scaffold to obtain 6-substituted carbocyclicnu- cleozide analogs as antiviral compounds. The synthesis was realized by a Mitsunobu reaction of the base with the corresponding bicyclo[2.2.0]heptane intermediate, and then the nucleoside analogs were obtained by substitution of the 6-chlorime with selected pharmaceutically accepted amines. A molecular docking study of the compounds on influenza, HSV and low active coronavirus was re- alized. Experimental screening of the compounds on the same viruses is being developed and soon Citation: Tănase, C.I.; Drăghici, C.; will be finished. -

Rash with Entecavir - Case Report Xiong Khee Cheong1, Zhiqin Wong2*, Norazirah Md Nor3 and Bang Rom Lee4
Cheong et al. BMC Gastroenterology (2020) 20:305 https://doi.org/10.1186/s12876-020-01452-3 CASE REPORT Open Access “Black box warning” rash with entecavir - case report Xiong Khee Cheong1, Zhiqin Wong2*, Norazirah Md Nor3 and Bang Rom Lee4 Abstract Background: Hepatitis B infection is a significant worldwide health issue, predispose to the development of liver cirrhosis and hepatocellular carcinoma. Entecavir is a potent oral antiviral agent of high genetic barrier for the treatment of chronic hepatitis B infection. Cutaneous adverse reaction associated with entecavir has rarely been reported in literature. As our knowledge, this case was the first case reported on entecavir induced lichenoid drug eruption. Case presentation: 55 year old gentlemen presented with generalised pruritic erythematous rash on trunk and extremities. Six weeks prior to his consultation, antiviral agent entecavir was commenced for his chronic hepatitis B infection. Skin biopsy revealed acanthosis and focal lymphocytes with moderate perivascular lymphocyte infiltration. Skin condition recovered completely after caesation of offending drug and short course of oral corticosteroids. Conclusion: This case highlight the awareness of clinicians on the spectrum of cutaneous drug reaction related to entecavir therapy. Keywords: Drug eruption, Entecavir, Hepatitis B Background Case presentation Hepatitis B infection is a global health issue, contribut- A 55 years old gentlemen, background of chronic ing to approximately 887,000 deaths in 2015 due to he- hepatitis B (treatment naïve), was referred for 2 patocellular carcinoma and liver cirrhosis [1, 2]. weeks of generalised itchy erythematous rash. His Entecavir is a nucleoside analogue reverse transcriptase other medical illnesses were ischemic heart disease, inhibitor that is widely used in the treatment of chronic chronic kidney disease stage 3, diabetes mellitus and hepatitis B (HBV) infection. -

Recent Advances in Antiviral Therapy J Clin Pathol: First Published As 10.1136/Jcp.52.2.89 on 1 February 1999
J Clin Pathol 1999;52:89–94 89 Recent advances in antiviral therapy J Clin Pathol: first published as 10.1136/jcp.52.2.89 on 1 February 1999. Downloaded from Derek Kinchington Abstract indicated that using a combination of drugs In the early 1980s many institutions in might overcome this problem. The only Britain were seriously considering available drugs during the late 1980s were two whether there was a need for specialist other nucleotide reverse transcriptase inhibi- departments of virology. The arrival of tors (NRTI) which also targeted HIV reverse HIV changed that perception and since transcriptase (HIV-RT): 2',3'-dideoxycytidine then virology and antiviral chemotherapy (ddC) and 2',3'-dideoxyinosine (ddI).56 In have become two very active areas of bio- vitro combination studies gave surprising medical research. Cloning and sequencing results: those viruses that became highly resist- have provided tools to identify viral en- ant to ZDV remained sensitive to both ddC zymes and have brought the day of the and ddI.7 Furthermore, neither cross resistance “designer drug” nearer to reality. At the nor interference between the drugs was an other end of the spectrum of drug discov- issue, and subsequent clinical experience ery, huge numbers of compounds for showed that patients benefited when these two screening can now be generated by combi- compounds were used in combination with natorial chemistry. The impetus to find ZDV.8 It was also found by in vitro studies that drugs eVective against HIV has also virus isolated from patients on long term ZDV stimulated research into novel treatments monotherapy had become insensitive to ZDV, for other virus infections including her- but regained sensitivity when these patients pesvirus, respiratory infections, and were switched to ddI monotherapy. -

Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection
pharmaceuticals Review Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection Isabella Zanella 1,2,* , Daniela Zizioli 1, Francesco Castelli 3 and Eugenia Quiros-Roldan 3 1 Department of Molecular and Translational Medicine, University of Brescia, 25123 Brescia, Italy; [email protected] 2 Clinical Chemistry Laboratory, Cytogenetics and Molecular Genetics Section, Diagnostic Department, ASST Spedali Civili di Brescia, Piazzale Spedali Civili 1, 25123 Brescia, Italy 3 University Department of Infectious and Tropical Diseases, University of Brescia and ASST Spedali Civili di Brescia, Piazzale Spedali Civili 1, 25123 Brescia, Italy; [email protected] (F.C.); [email protected] (E.Q.-R.) * Correspondence: [email protected]; Tel.: +39-030-399-6806 Abstract: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is spreading worldwide with different clinical manifestations. Age and comorbidities may explain severity in critical cases and people living with human immunodeficiency virus (HIV) might be at particularly high risk for severe progression. Nonetheless, current data, although sometimes contradictory, do not confirm higher morbidity, risk of more severe COVID-19 or higher mortality in HIV-infected people with complete access to antiretroviral therapy (ART). A possible protective role of ART has been hypothesized to explain these observations. Anti-viral drugs used to treat HIV infection have been repurposed for COVID-19 treatment; this is also based on previous studies on severe acute respiratory syndrome virus (SARS-CoV) and Middle East respiratory syndrome virus (MERS-CoV). Among Citation: Zanella, I.; Zizioli, D.; them, lopinavir/ritonavir, an inhibitor of viral protease, was extensively used early in the pandemic Castelli, F.; Quiros-Roldan, E. -

Investigating Synergy Between Ribonucleotide Reductase Inhibitors and Cmv Antivirals
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2012 INVESTIGATING SYNERGY BETWEEN RIBONUCLEOTIDE REDUCTASE INHIBITORS AND CMV ANTIVIRALS Sukhada Bhave Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Medicine and Health Sciences Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2838 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. © Sukhada Milind Bhave August 2012 All Rights Reserved INVESTIGATING SYNERGY BETWEEN RIBONUCLEOTIDE REDUCTASE INHIBITORS AND CMV ANTIVIRALS A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by Sukhada Milind Bhave Bachelor of Pharmacy, Bombay College of Pharmacy, India, 2010 Major Director: Michael McVoy, Ph.D. Professor, Department of Microbiology and Immunology Virginia Commonwealth University Richmond, Virginia August 2012 ACKNOWLEDGEMENTS It is a pleasure to thank everyone who made this thesis possible. First, I would like to thank Dr. McVoy for accepting me as a graduate student in his lab and for his guidance and support through this project. I would also like to thank the other members of the McVoy lab: Frances Saccoccio, Anne Sauer, Xiao Cui, Ronzo Lee, Ben Wang, and Sabrina Prescott for sharing their expertise and helping me through various steps in the project. All of them indeed create a very cheerful and pleasant atmosphere in the lab. -

The Evolution of Antiviral Therapy for External Ocular Viral Infections Over Twenty-Five Years
Cornea 19(5): 673–680, 2000. © 2000 Lippincott Williams & Wilkins, Inc., Philadelphia The Evolution of Antiviral Therapy for External Ocular Viral Infections Over Twenty-five Years Y. Jerold Gordon, M.D. Purpose. To review the past 25 years of the evolution of antiviral levels on the corneal surface. By 1975, IDU was the treatment of therapy for the treatment of common external ocular viral infec- choice for herpetic epithelial keratitis worldwide. However, the tions (herpes simplex virus type 1, varicella-zoster virus, and ad- shortcomings of topical IDU therapy: significant toxicity (super- enovirus). Methods. A broad-based literature review in the fields ficial punctate keratitis, chemical conjunctivitis, and punctal oc- of virology, antiviral research, and ophthalmology will be carried clusion) and frequent hypersensitivity reactions, limited its useful- out. The pathogenesis of the major external ocular viral infections ness. As an insoluble agent, it had no therapeutic effect on herpetic and history of antiviral development will be cited. Important con- ceptual breakthroughs as well as historical landmarks will be em- stromal keratitis or iritis. Today, IDU is used infrequently in de- phasized. Results. The successful development of effective anti- veloped nations because it has been replaced by better drugs. How- virals to treat the most common external ocular viral infections ever, it remains effective and is used in developing nations where have dramatically reduced morbidity and sight loss. The immune cost issues are critical. pathogenesis of herpetic stromal keratitis is better understood. In the mid-1970s, several promising strategies to treat HSV Conclusions. Remarkable progress in the development of antiviral epithelial keratitis failed. -

Drug Interaction Studies on New Drug Applications: Current Situations and Regulatory Views in Japan
Drug Metab. Pharmacokinet. 25 (1): 3–15 (2010). Review Drug Interaction Studies on New Drug Applications: Current Situations and Regulatory Views in Japan Naomi NAGAI* Office of New Drug IV, Pharmaceuticals and Medical Devices Agency (PMDA), Tokyo, Japan Full text of this paper is available at http://www.jstage.jst.go.jp/browse/dmpk Summary: Drug interaction studies on new drug applications (NDAs) for new molecular entities (NMEs) ap- proved in Japan between 1997 and 2008 are examined in the Pharmaceuticals and Medical Devices Agency (PMDA). The situations of drug interaction studies in NDAs have changed over the past 12 years, especially in metabolizing enzyme and transporter-based drug interactions. Materials and approaches to study drug- metabolizing enzyme-based drug interactions have improved, and become more rational based on mechanis- tic theory and new technologies. On the basis of incremental evidence of transporter roles in human phar- macokinetics, transporter-based drug interactions have been increasingly studied during drug development and submitted in recent NDAs. Some recently approved NMEs include transporter-based drug interaction in- formation in their package inserts (PIs). The regulatory document ``Methods of Drug Interaction Studies,'' in addition to recent advances in science and technology, has also contributed to plan and evaluation of drug in- teraction studies in recent new drug development. This review summarizes current situations and further dis- cussionpointsondruginteractionstudiesinNDAsinJapan. Keywords: -

Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2
viruses Review Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2 Lauren A. Sadowski 1,†, Rista Upadhyay 1,2,†, Zachary W. Greeley 1,‡ and Barry J. Margulies 1,3,* 1 Towson University Herpes Virus Lab, Department of Biological Sciences, Towson University, Towson, MD 21252, USA; [email protected] (L.A.S.); [email protected] (R.U.); [email protected] (Z.W.G.) 2 Towson University Department of Chemistry, Towson, MD 21252, USA 3 Molecular Biology, Biochemistry, and Bioinformatics Program, Towson University, Towson, MD 21252, USA * Correspondence: [email protected] † Authors contributed equally to this manuscript. ‡ Current address: Becton-Dickinson, Sparks, MD 21152, USA. Abstract: Herpes simplex viruses-1 and -2 (HSV-1 and -2) are two of the three human alphaher- pesviruses that cause infections worldwide. Since both viruses can be acquired in the absence of visible signs and symptoms, yet still result in lifelong infection, it is imperative that we provide interventions to keep them at bay, especially in immunocompromised patients. While numerous experimental vaccines are under consideration, current intervention consists solely of antiviral chemotherapeutic agents. This review explores all of the clinically approved drugs used to prevent the worst sequelae of recurrent outbreaks by these viruses. Keywords: acyclovir; ganciclovir; cidofovir; vidarabine; foscarnet; amenamevir; docosanol; nelfi- navir; HSV-1; HSV-2 Citation: Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections 1. Introduction with Herpes Simplex Viruses-1 and -2. The world of anti-herpes simplex (anti-HSV) agents took flight in 1962 with the FDA Viruses 2021, 13, 1228.