Microbial Diversity of Co-Occurring Heterotrophs in Cultures of Marine Picocyanobacteria Sean M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fermentation and Anaerobic Decomposition in a Hot Spring
Fermentation and anaerobic decomposition in a hot spring microbial mat by Karen Leigh Anderson A thesis submitted in partial fulfillment of requirements for the degree of Master of Science in Microbiology Montana State University © Copyright by Karen Leigh Anderson (1984) Abstract: Fermentation was investigated in a low sulfate hot spring microbial mat (Octopus Spring) according to current models on anaerobic decomposition. The mat was studied to determine what fermentation products accumulated, where in the mat they accumulated, and what factors affected their accumulation. Mat samples were incubated under dark anaerobic conditions to measure accumulation of fermentation products. Acetate and propionate (ca. 3:1) were the major products to accumulate in a 55°,C mat. Other products accumulated to a much lesser extent. Incubation of mat samples of varying thickness showed that fermentation occurred in the top 4mm of the mat. This has interesting implications for fermentative organisms in the mat due to the diurnal changes in mat oxygen concentrations. Fermentation measured in mat samples collected at various temperatures (50°,-70°C) showed acetate and propionate to be the major accumulation products. According to the interspecies hydrogen transfer model, the hydrogen concentration in a system affects the types of fermentation products produced. At a 65° C site, with natural high hydrogen levels, and at a 55°C site, with active methanogenesis, fermentation product accumulation was compared. There was a greater ratio of reduced fermentation products to acetate, with the exception of propionate, at 65°C. Ethanol accumulated at the 65°C site, as did lactate, though to a lesser extent. -

Principles of Ecology Section ●2 Flow of Energy in an Ecosystem
chapter 2 Principles of Ecology section ●2 Flow of Energy in an Ecosystem -!). )DEA Before You Read Autotrophs capture energy, making it available for all If a pet had to survive without your care, how would its diet members of a food web. change? Write your ideas on the lines below. Read about how What You’ll Learn organisms get food and energy in their environment. ■ the fl ow of energy through an ecosystem ■ food chains, food webs, and pyramid models 3TUDY#OACH Read to Learn Make Flash Cards Make a fl ash card for each question Energy in an Ecosystem heading in this section. On the One way to study the interactions within an ecosystem is to back of the fl ash card, write the trace how energy fl ows through the system. All organisms are answer to the question. Use the classifi ed by the way they obtain energy. fl ash cards to review what you have learned. How do autotrophs obtain energy? All green plants and other organisms that produce their own food are the primary producers of food in an ecosystem. They are called autotrophs. An autotroph (AW tuh trohf) is an organism that captures energy from sunlight or inorganic substances to produce food. Autotrophs make energy available for all other organisms in the ecosystem. How do heterotrophs differ from autotrophs? A heterotroph (HE tuh roh trohf), also called a consumer, is an organism that obtains energy by consuming other organisms. A heterotroph that consumes only plants is an herbivore (HUR buh vor). Cows, rabbits, and grasshoppers are herbivores. -

7.014 Lectures 16 &17: the Biosphere & Carbon and Energy Metabolism
MIT Department of Biology 7.014 Introductory Biology, Spring 2005 7.014 Lectures 16 &17: The Biosphere & Carbon and Energy Metabolism Simplified Summary of Microbial Metabolism The metabolism of different types of organisms drives the biogeochemical cycles of the biosphere. Balanced oxidation and reduction reactions keep the system from “running down”. All living organisms can be ordered into two groups1, autotrophs and heterotrophs, according to what they use as their carbon source. Within these groups the metabolism of organisms can be further classified according to their source of energy and electrons. Autotrophs: Those organisms get their energy from light (photoautotrophs) or reduced inorganic compounds (chemoautotrophs), and their carbon from CO2 through one of the following processes: Photosynthesis (aerobic) — Light energy used to reduce CO2 to organic carbon using H2O as a source of electrons. O2 evolved from splitting H2O. (Plants, algae, cyanobacteria) Bacterial Photosynthesis (anaerobic) — Light energy used to reduce CO2 to organic carbon (same as photosynthesis). H2S is used as the electron donor instead of H2O. (e.g. purple sulfur bacteria) Chemosynthesis (aerobic) — Energy from the oxidation of inorganic molecules is used to reduce CO2 to organic carbon (bacteria only). -2 e.g. sulfur oxidizing bacteria H2S → S → SO4 + - • nitrifying bacteria NH4 → NO2 → NO3 iron oxidizing bacteria Fe+2 → Fe+3 methane oxidizing bacteria (methanotrophs) CH4 → CO2 Heterotrophs: These organisms get their energy and carbon from organic compounds (supplied by autotrophs through the food web) through one or more of the following processes: Aerobic Respiration (aerobic) ⎯ Oxidation of organic compounds to CO2 and H2O, yielding energy for biological work. -
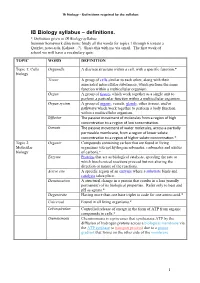
IB Biology Syllabus – Definitions
IB Biology – Definitions required by the syllabus IB Biology syllabus – definitions. * Definition given in IB Biology syllabus Summer homework directions: Study all the words for topics 1 through 6 (create a Quizlet, notecards, Kahoot…?). Share this with me via email. The first week of school we will have a vocabulary quiz. TOPIC WORD DEFINITION Topic 1: Cells Organelle A discrete structure within a cell, with a specific function.* biology Tissue A group of cells similar to each other, along with their associated intercellular substances, which perform the same function within a multicellular organism. Organ A group of tissues which work together as a single unit to perform a particular function within a multicellular organism. Organ system A group of organs, vessels, glands, other tissues, and/or pathways which work together to perform a body function within a multicellular organism. Diffusion The passive movement of molecules from a region of high concentration to a region of low concentration. Osmosis The passive movement of water molecules, across a partially permeable membrane, from a region of lower solute concentration to a region of higher solute concentration.* Topic 2: Organic Compounds containing carbon that are found in living Molecular organisms (except hydrogencarbonates, carbonates and oxides biology of carbon).* Enzyme Proteins that act as biological catalysts, speeding the rate at which biochemical reactions proceed but not altering the direction or nature of the reactions. Active site A specific region of an enzyme where a substrate binds and catalysis takes place. Denaturation A structural change in a protein that results in a loss (usually permanent) of its biological properties. -

The Heterotroph Hypothesis for the Evolution of the Basic Metabolic Processes an Explanatory Scenario That Helps to Show the Logic of This Hypothesis
The Heterotroph Hypothesis for the Evolution of the Basic Metabolic Processes An explanatory scenario that helps to show the logic of this hypothesis The Heterotroph Hypothesis is the proposal that the first living organism was a HETEROTROPH. Heterotrophs are organisms that obtain their energy by feeding on others (or on organic compounds) Hetero = other troph = to feed Before there were other organisms, they would feed on surrounding “left-overs” of their origin. The simplest process we know of today by which organisms do this is called FERMENTATION There is evidence that the pre-life atmosphere and water environment was a reducing one, (no oxygen). Evidence: There are no iron oxides (rust-red rocks) in unexposed deposits of that age. The waste products of Fermentation are ethyl alcohol (ethanol) and carbon dioxide (CO2) The main useful product of Fermentation is the chemical energy extracted from the chemical bonds of organic compounds, and used to produce energy-rich packets of ATP, available for any and all biochemical processes requiring energy. Some of that energy was used to build a variety of chemicals, some of which were pigmented. Evidence: Experiments in which a variety of organic compounds are placed in a sterile environment with energy sources randomly produce substances of various colors. As the thick clouds of water vapor cleared over this primitive earth, the energy of sunlight filtered through. Organisms with pigments that could absorb light and transform that energy into a chemical form that could be used for biochemical processes (leading to growth and reproduction) would spread and become more efficient. -

Biology Sol Review: a Quick Overview
BIOLOGY SOL REVIEW: A QUICK OVERVIEW Useful Web sites 1. Released SOL Tests, Interactive, Provides Score Who is guilty? Read the DNA Fingerprint http://www.solpass.org/hs.htm#biology 2. Flashcard Review http://www.studystack.com/flashcard-38612 3. Really good interactive biology review questions http://www.bedford.k12.va.us/srhs/sol/biology1.htm 4. Review with a few or many questions. Select # of questions& biology http://education.jlab.org/solquiz/index.html Ecology Commensalism 1 benefits, 0 harmed Orchid in a tree Odds & Ends: Mutualism 2 benefit, 0 harm Bacterial in human intestine Parasitism 1 benefit, 1 harmed Tapeworm Ingest: to consume Producer Autotroph Plant, photosynthetic Splice: to join together bacteria or protist Optimum: best Consumer Heterotroph Must eat Aquatic: water Decomposer Breaking down dead Fungi or bacteria growing Terrestrial: land organisms on a dead tree Scavenger/detritivore Eats dead organisms Buzzard eating a dead skunk Herbivore Eats plants aphids Carnivore Eats meat Dog Omnivore Eats plants & meat Humans Primary consumer Always a herbivore Pioneer 1st photosynthetic organism Ex. Moss or lichens to colonize barren area Secondary consumer Eats primary consumer May also be an omnivore Tertiary consumer Eats secondary consumer May be an omnivore Primary succession New life Melting glacier, new island Secondary succession New life where there was Mt. St. Helens, forest fire life before Punctuated No change followed by equilibrium short periods of abrupt Homeostasis: maintaining a constant internal (evolution) -
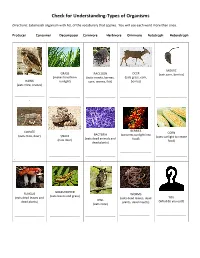
Check for Understanding-Types of Organisms
Check for Understanding-Types of Organisms Directions: Label each organism with ALL of the vocabulary that applies. You will use each word more than once. Producer Consumer Decomposer Carnivore Herbivore Omnivore Autotroph Heterotroph MOUSE GRASS RACCOON DEER (eats corn, berries) (makes food from (eats insects, berries, (eats grass, corn, HAWK sunlight) corn, worms, fish) berries) (eats mice, snakes) ------------------------------ ------------------------------- ------------------------------ ------------------------------- ------------------------------- - BERRIES COYOTE CORN BACTERIA (converts sunlight into (eats mice, deer) SNAKE (uses sunlight to create (eats dead animals and food) (eats mice) food) dead plants) ------------------------------- ------------------------------ ------------------------------- ------------------------------ ------------------------------- - - GRASSHOPPER FUNGUS WORMS (eats leaves and grass) (eats dead leaves and (eats dead leaves, dead YOU OWL dead plants) plants, dead insects) (What do you eat?) (eats mice) ------------------------------ ------------------------------ ------------------------------ ------------------------------ ------------------------------ - MOUSE GRASS RACCOON DEER (eats corn, berries) (makes food from (eats insects, berries, (eats grass, corn, HAWK sunlight) corn, worms, fish) berries) (eats mice, snakes) Consumer Producer Consumer Consumer Consumer Carnivore Autotroph Omnivore Herbivore Herbivore Heterotroph Heterotroph Heterotroph Heterotroph BERRIES COYOTE CORN BACTERIA (converts -

Igem Microbiology Intro
iGEM Microbiology Intro Prof. Sebasan Maerkl Index • Prokaryoc Structure and Funcon – Cell components – Chemotaxis and flagellar – Living magnets • Nutrion and Growth – Growth requirements – Plang techniques – Growth curve – Chemostats / turbidostats • Microbial Genecs – Recombinaon – Transposons – Plasmids – Conjugaon – Transformaon – Transducon Prokaryoc Cell Structure • Plasma Membrane • Gas vacuole: buoyancy • Ribosomes: protein synthesis • Inclusion Bodies: storage of C, P, etc • Nucleoid: chromosomal DNA • Periplasmic space: hydrolyc and binding proteins • Cell Wall: mechanical rigidity • Capsules: resistance to phagocytosis; adhesion • Fimbriae and pili: aachments and mang • Flagella: movement • Endospore: survival mechanism 10.1016/j.cell.2008.11.016 Morphology and Classificaon Gram-Positive Gram-Negative Flagella and Chemotaxis • Tumble: – CheA autophosphorylates if MCP is not bound – CheA then phosphorylates CheY, which causes a tumble (CheYp is rapidly degraded to CheY by CheZ • Run: – Aractant binds to MCP causing a stearic shi, liming autophosphorylaon of CheA – CheYp levels drop, causing CCW rotaon • Reseng: – CheR methylates MCPs causing increased CheA autophosphorylaon – This causes increases in CheYp and CheBp, which lead to CW rotaon and de‐methylaon of MCPs Living Magnets (Magnetotacc Bacteria) • Magnetosomes consist of magnec iron mineral crystals made from: Fe3O4, greigite, or Fe3S4 • Magnetosomes align the bacteria in the geomagnec field, allowing for directed taxis Microbial Metabolism • Prototrophs: can thrive on minimal -

Glossary.Pdf
Glossary Pronunciation Key accessory fruit A fruit, or assemblage of fruits, adaptation Inherited characteristic of an organ- Pronounce in which the fleshy parts are derived largely or ism that enhances its survival and reproduc- a- as in ace entirely from tissues other than the ovary. tion in a specific environment. – Glossary Ј Ј a/ah ash acclimatization (uh-klı¯ -muh-tı¯-za -shun) adaptive immunity A vertebrate-specific Physiological adjustment to a change in an defense that is mediated by B lymphocytes ch chose environmental factor. (B cells) and T lymphocytes (T cells). It e¯ meet acetyl CoA Acetyl coenzyme A; the entry com- exhibits specificity, memory, and self-nonself e/eh bet pound for the citric acid cycle in cellular respi- recognition. Also called acquired immunity. g game ration, formed from a fragment of pyruvate adaptive radiation Period of evolutionary change ı¯ ice attached to a coenzyme. in which groups of organisms form many new i hit acetylcholine (asЈ-uh-til-ko–Ј-le¯n) One of the species whose adaptations allow them to fill dif- ks box most common neurotransmitters; functions by ferent ecological roles in their communities. kw quick binding to receptors and altering the perme- addition rule A rule of probability stating that ng song ability of the postsynaptic membrane to specific the probability of any one of two or more mu- o- robe ions, either depolarizing or hyperpolarizing the tually exclusive events occurring can be deter- membrane. mined by adding their individual probabilities. o ox acid A substance that increases the hydrogen ion adenosine triphosphate See ATP (adenosine oy boy concentration of a solution. -

General Introduction
UvA-DARE (Digital Academic Repository) Viral lysis of marine microbes in relation to vertical stratification Mojica, K.D.A. Publication date 2015 Document Version Final published version Link to publication Citation for published version (APA): Mojica, K. D. A. (2015). Viral lysis of marine microbes in relation to vertical stratification. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:25 Sep 2021 Chapter 1 General Introduction 35502_Mojica.indd 9 06-08-15 22:51 Chapter 1 Viruses are the most abundant biological entities in the oceans and are important mortality agents of heterotrophic and autotrophic microbial populations. These microbial hosts are numerically dominant and constitute the largest percentage (>90%) of living biomass in the ocean. Collectively microorganisms manage the pools and fluxes of energy and nutrients in the ocean. -

Odyssey High School Biology Vocabulary
ODYSSEY HIGH SCHOOL BIOLOGY VOCABULARY These are the vocabulary words and definitions used throughout the Biology course.They are listed in alphabetical order. Vocabulary Word Definition abiotic physical, or nonliving, factor that shapes an ecosystem abiotic factor physical, or nonliving, part of an ecosystem accurate the closeness of a measurement to the actual value acoelomate having no body cavity acoelomic lacking a body cavity acquired traits traits that develop as a result of an organism's experiences in the world the energy needed to start a reaction by exciting a stable molecule and activation energy making it a reactive molecule the energy-requiring process of moving or pumping a substance across the active transport plasma membrane against an electrical or concentration gradient (moving from low to high concentration) adapt to change to suit a new purpose a variation in a plant or animal that increases it chance of survival in its adaptation environment evolutionary divergence of members of a single phyletic line into many adaptive radiation different niches adenine purine base that pairs with thymine in DNA or uracil in RNA a small gland that produces steroid hormones, adrenaline and adrenal gland nonadrenaline, which helps control heart rate, blood pressure adventitious roots roots growing from an unusual spot, like the stem cellular process of producing energy in the form of ATP and carbon dioxide aerobic respiration from food (glucose) and oxygen; occurs in the mitochondria air the mixture of gases that make up the atmosphere -

Life Science GLOSSARY
Life Science GLOSSARY angiosperm (AN-jee-uh-sPURM) A A plant that has flowers and produces seeds en- abiotic factor (AY-by-AHT-i hk) closed in fruit. (p. 379) A nonliving physical or chemical part of an ecosystem. (p. 474) Animalia (AN-U h-MAL-yu h) Part of a classification system that divides all living active transport things into six kingdoms. Kingdom Animalia includes The process of using energy to move materials multicellular organisms, from humans and lions to through a membrane. (p. 60) insects and microbes, that rely on food for energy. (p. 223) adaptation A characteristic, a behavior, or any inherited trait that antibiotic makes a species able to survive and reproduce in a A medicine that can block the growth and reproduc- particular environment. (p. xxxvii) tion of bacteria. (p. 65 7) addiction antibody A physical or psychological need for a habit-forming A protein produced by some white blood cells to at- substance, such as alcohol or drugs. (p. 722) tack specific foreign materials. (p. 651) adolescence (Ao- u hi-EHS-uhns) antigen The stage of life from the time a human body begins A particular substance that the body recognizes as to mature sexually to adulthood. (p. 711) foreign and that stimulates a response. (p. 654) dulthood appendicular skeleton (AP-u hn-DIH K-yu h-lu hr) he stage of life that begins once a human body com- The bones of the skeleton that function to allow letes movement, such as arm and leg bones. (p. 592) its growth and reaches sexual maturity.