Covalently-Linked Complexes and Methods for Enhanced Cytotoxicity and Imaging
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
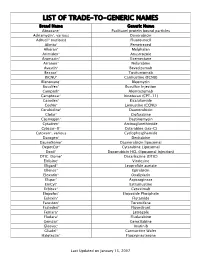
Trade-To-Generic Names
LIST OF TRADE-TO-GENERIC NAMES Brand Name Generic Name Abraxane® Paclitaxel protein bound particles Adriamycin®, various Doxorubicin Adrucil® (various) Fluorouracil Alimta® Pemetrexed Alkeran® Melphalan Arimidex® Anastrozole Aromasin® Exemestane Arranon® Nelarabine Avastin® Bevacizumab Bexxar® Tositumomab BiCNU® Carmustine (BCNU) Blenoxane® Bleomycin Busulfex® Busulfan Injection Campath® Alemtuzumab Camptosar® Irinotecan (CPT-11) Casodex® Bicalutamide CeeNu® Lomustine (CCNU) Cerubidine® Daunorubicin Clolar® Clofarabine Cosmegen® Dactinomycin Cytadren® Aminoglutethimide Cytosar-U® Cytarabine (ara-C) Cytoxan®, various Cyclophosphamide Dacogen® Decitabine DaunoXome® Daunorubicin liposomal DepotCyt® Cytarabine Liposomal Doxil® Doxorubicin HCL (liposomal injection) DTIC-Dome® Dacarbazine (DTIC) Eldisine® Vindesine Eligard® Leuprolide acetate Ellence® Epirubicin Eloxatin® Oxaliplatin Elspar® Asparaginase EmCyt® Estramustine Erbitux® Cetuximab Etopofos® Etoposide Phosphate Eulexin® Flutamide Fareston® Toremifene Faslodex® Fluvestrant Femara® Letrozole Fludara® Fludarabine Gemzar® Gemcitabine Gleevec® Imatinib Gliadel® Carmustine Wafer Halotestin® Fluoxymesterone Last Updated on January 15, 2007 Brand Name Generic Name Herceptin® Trastuzumab Hexalen® Altretamine Hycamtin® Topotecan Hydrea® Hydroxyurea Idamycin® Idarubicin Ifex® Ifosfamide Intron A® Interferon alfa-2b Iressa® Gefitinib Leukeran® Chlorambucil Leukine® Sargramostim Leustatin® Cladribine Lupron depot® Leuprolide acetate depot Lupron® Leuprolide acetate Matulane® Procarbazine Megace® -

Phenotype Microarrays Panels PM-M1 to PM-M14
Phenotype MicroArrays™ Panels PM-M1 to PM-M14 for Phenotypic Characterization of Mammalian Cells Assays: Energy Metabolism Pathways Ion and Hormone Effects on Cells Sensitivity to Anti-Cancer Agents and for Optimizing Culture Conditions for Mammalian Cells PRODUCT DESCRIPTIONS AND INSTRUCTIONS FOR USE PM-M1 Cat. #13101 PM-M2 Cat. #13102 PM-M3 Cat. #13103 PM-M4 Cat. #13104 PM-M5 Cat. #13105 PM-M6 Cat. #13106 PM-M7 Cat. #13107 PM-M8 Cat. #13108 PM-M11 Cat. #13111 PM-M12 Cat. #13112 PM-M13 Cat. #13113 PM-M14 Cat. #13114 © 2016 Biolog, Inc. All rights reserved Printed in the United States of America 00P 134 Rev F February 2020 - 1 - CONTENTS I. Introduction ...................................................................................................... 2 a. Overview ................................................................................................... 2 b. Background ............................................................................................... 2 c. Uses ........................................................................................................... 2 d. Advantages ................................................................................................ 3 II. Product Description, PM-M1 to M4 ................................................................ 3 III. Protocols, PM-M1 to M4 ................................................................................. 7 a. Materials Required .................................................................................... 7 b. Determination -

NINDS Custom Collection II
ACACETIN ACEBUTOLOL HYDROCHLORIDE ACECLIDINE HYDROCHLORIDE ACEMETACIN ACETAMINOPHEN ACETAMINOSALOL ACETANILIDE ACETARSOL ACETAZOLAMIDE ACETOHYDROXAMIC ACID ACETRIAZOIC ACID ACETYL TYROSINE ETHYL ESTER ACETYLCARNITINE ACETYLCHOLINE ACETYLCYSTEINE ACETYLGLUCOSAMINE ACETYLGLUTAMIC ACID ACETYL-L-LEUCINE ACETYLPHENYLALANINE ACETYLSEROTONIN ACETYLTRYPTOPHAN ACEXAMIC ACID ACIVICIN ACLACINOMYCIN A1 ACONITINE ACRIFLAVINIUM HYDROCHLORIDE ACRISORCIN ACTINONIN ACYCLOVIR ADENOSINE PHOSPHATE ADENOSINE ADRENALINE BITARTRATE AESCULIN AJMALINE AKLAVINE HYDROCHLORIDE ALANYL-dl-LEUCINE ALANYL-dl-PHENYLALANINE ALAPROCLATE ALBENDAZOLE ALBUTEROL ALEXIDINE HYDROCHLORIDE ALLANTOIN ALLOPURINOL ALMOTRIPTAN ALOIN ALPRENOLOL ALTRETAMINE ALVERINE CITRATE AMANTADINE HYDROCHLORIDE AMBROXOL HYDROCHLORIDE AMCINONIDE AMIKACIN SULFATE AMILORIDE HYDROCHLORIDE 3-AMINOBENZAMIDE gamma-AMINOBUTYRIC ACID AMINOCAPROIC ACID N- (2-AMINOETHYL)-4-CHLOROBENZAMIDE (RO-16-6491) AMINOGLUTETHIMIDE AMINOHIPPURIC ACID AMINOHYDROXYBUTYRIC ACID AMINOLEVULINIC ACID HYDROCHLORIDE AMINOPHENAZONE 3-AMINOPROPANESULPHONIC ACID AMINOPYRIDINE 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE AMINOTHIAZOLE AMIODARONE HYDROCHLORIDE AMIPRILOSE AMITRIPTYLINE HYDROCHLORIDE AMLODIPINE BESYLATE AMODIAQUINE DIHYDROCHLORIDE AMOXEPINE AMOXICILLIN AMPICILLIN SODIUM AMPROLIUM AMRINONE AMYGDALIN ANABASAMINE HYDROCHLORIDE ANABASINE HYDROCHLORIDE ANCITABINE HYDROCHLORIDE ANDROSTERONE SODIUM SULFATE ANIRACETAM ANISINDIONE ANISODAMINE ANISOMYCIN ANTAZOLINE PHOSPHATE ANTHRALIN ANTIMYCIN A (A1 shown) ANTIPYRINE APHYLLIC -

Electrification at Water–Hydrophobe Interfaces
Electrification at water–hydrophobe interfaces Item Type Article Authors Nauruzbayeva, Jamilya; Sun, Zhonghao; Gallo Junior, Adair; Ibrahim, Mahmoud; Santamarina, Carlos; Mishra, Himanshu Citation Nauruzbayeva, J., Sun, Z., Gallo, A., Ibrahim, M., Santamarina, J. C., & Mishra, H. (2020). Electrification at water–hydrophobe interfaces. Nature Communications, 11(1). doi:10.1038/ s41467-020-19054-8 Eprint version Publisher's Version/PDF DOI 10.1038/s41467-020-19054-8 Publisher Springer Nature Journal Nature Communications Rights This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. Download date 05/10/2021 16:06:59 Item License https://creativecommons.org/licenses/by/4.0 Link to Item http://hdl.handle.net/10754/665657 ARTICLE https://doi.org/10.1038/s41467-020-19054-8 OPEN Electrification at water–hydrophobe interfaces Jamilya Nauruzbayeva1,3, Zhonghao Sun 2,3, Adair Gallo Jr.1,3, Mahmoud Ibrahim1, J. Carlos Santamarina 2 & ✉ Himanshu Mishra 1 The mechanisms leading to the electrification of water when it comes in contact with hydrophobic surfaces remains a research frontier in chemical science. -

A Comparison Between Triplet and Doublet Chemotherapy in Improving
Guo et al. BMC Cancer (2019) 19:1125 https://doi.org/10.1186/s12885-019-6294-9 RESEARCH ARTICLE Open Access A comparison between triplet and doublet chemotherapy in improving the survival of patients with advanced gastric cancer: a systematic review and meta-analysis Xinjian Guo1, Fuxing Zhao1, Xinfu Ma1, Guoshuang Shen1, Dengfeng Ren1, Fangchao Zheng1,2,3, Feng Du4, Ziyi Wang1, Raees Ahmad1, Xinyue Yuan1, Junhui Zhao1* and Jiuda Zhao1* Abstract Background: Chemotherapy can improve the survival of patients with advanced gastric cancer. However, whether triplet chemotherapy can further improve the survival of patients with advanced gastric cancer compared with doublet chemotherapy remains controversial. This study reviewed and updated all published and eligible randomized controlled trials (RCTs) to compare the efficacy, prognosis, and toxicity of triplet chemotherapy with doublet chemotherapy in patients with advanced gastric cancer. Methods: RCTs on first-line chemotherapy in advanced gastric cancer on PubMed, Embase, and the Cochrane Register of Controlled Trials and all abstracts from the annual meetings of the European Society for Medical Oncology (ESMO) and the American Society of Clinical Oncology conferences up to October 2018 were searched. The primary outcome was overall survival, while the secondary outcomes were progression-free survival (PFS), time to progress (TTP), objective response rate (ORR), and toxicity. Results: Our analysis included 23 RCTs involving 4540 patients and 8 types of triplet and doublet chemotherapy regimens, and systematic review and meta-analysis revealed that triplet chemotherapy was superior compared with doublet chemotherapy in terms of improving median OS (HR = 0.92; 95% CI, 0.86–0.98; P = 0.02) and PFS (HR = 0.82; 95% CI, 0.69–0.97; P = 0.02) and TTP (HR = 0.92; 95% CI, 0.86–0.98; P = 0.02) and ORR (OR = 1.21; 95% CI, 1.12–1.31; P < 0.0001) among overall populations. -

Affinity of Small-Molecule Solutes to Hydrophobic, Hydrophilic, and Chemically Patterned Interfaces in Aqueous Solution
Affinity of small-molecule solutes to hydrophobic, hydrophilic, and chemically patterned interfaces in aqueous solution Jacob I. Monroea, Sally Jiaoa, R. Justin Davisb, Dennis Robinson Browna, Lynn E. Katzb, and M. Scott Shella,1 aDepartment of Chemical Engineering, University of California, Santa Barbara, CA 93106; and bDepartment of Civil, Architectural and Environmental Engineering, University of Texas at Austin, Austin, TX 78712 Edited by Peter J. Rossky, Rice University, Houston, TX, and approved November 17, 2020 (received for review September 30, 2020) Performance of membranes for water purification is highly influ- However, a molecular understanding that links membrane enced by the interactions of solvated species with membrane surface chemistry to solute affinity and hence membrane func- surfaces, including surface adsorption of solutes upon fouling. tional properties remains incomplete, due in part to the complex Current efforts toward fouling-resistant membranes often pursue interplay among specific interactions (e.g., hydrogen bonds, surface hydrophilization, frequently motivated by macroscopic electrostatics, dispersion) and molecular morphology (e.g., sur- measures of hydrophilicity, because hydrophobicity is thought to face and polymer configurations) that are difficult to disentangle increase solute–surface affinity. While this heuristic has driven di- (11–14). Chemically heterogeneous surfaces are even less un- verse membrane functionalization strategies, here we build on derstood but can affect fouling in complex ways. -

Biological Physics
Biological Physics FYS4715 2020 Some questions Write, 5 min, read aloud the parts you want, publish anonymously, helpful for me • Why did you choose to follow FYS4715? • What do you expect to get out of the course? • What have you learnt previously that is relevant for the course? • What education background do you have? Themes this year • General: Processes on cellular and molecular scale • 1nm – 100 um • from atomic to macroscopic: stat mech • small scale: atomic forces, velocities, charges • large scale: surface tension, stress-strain, diffusion, osmosis, electrochemistry • Nelson ch 1-8, 12 + chosen topics • Projects: • Passive and active swimmers • Cellular Potts model (?) • Mechanobiology, effects of ultrasound on cells • Bioimpedance • Nerve impulses Chapter 1 • What is heat? • Entropy • 2nd law vs. order • Free energy • Osmosis • Physical models • For biologists: https://www.natur e.com/articles/d41 586-019-03960-z Biological Physics • In physics we make mathematical models and calculate stuff • What is the volume of one molecule of water? • Most famous model: ideal gas • equation of state: PV = nkBT • What is PV? • How much is kBT? pN forces nm distances Chapter 2: What’s inside cells • Biological question: How do organisms organize all the chemical processes? • Physical ideas • Compartmentalization • Active transport • Specific processes • Non-linearity -> switching in networks Polar – hydrophilic – water soluble Hydrophobe & amphiphile higher energy cost energy and entropy Important molecules • Important nitrogenous bases: Adenine, -

Influence of Hydrophobe Fraction Content on the Rheological Properties of Hydrosoluble Associative Polymers Obtained by Micellar Polymerization
http://www.e-polymers.org e-Polymers 2012, no. 009 ISSN 1618-7229 Influence of hydrophobe fraction content on the rheological properties of hydrosoluble associative polymers obtained by micellar polymerization Enrique J. Jiménez-Regalado,1* Elva B. Hernández-Flores2 1Centro de Investigación en Química Aplicada (CIQA) Blvd. Enrique Reyna #140, 25253, Saltillo, Coahuila, México; e-mail: [email protected] bCentro de Investigación en Química Aplicada (CIQA) Blvd. Enrique Reyna #140, 25253, Saltillo, Coahuila, México; e-mail: [email protected] (Received: 20 November, 2009; published: 22 January, 2012) Abstract: The synthesis, characterization and rheological properties in aqueous solutions of water-soluble associative polymers (AP’s) are reported. Polymer chains consisting of water-soluble polyacrylamides, hydrophobically modified with low amounts of N,N-dihexylacrylamide (1, 2, 3 and 4 mol%) were prepared via free radical micellar polymerization. The properties of these polymers, with respect to the concentration of hydrophobic groups, using steady-flow and oscillatory experiments were compared. An increase of relaxation time (TR) and modulus plateau (G0) was observed in all samples studied. Two different regimes can be clearly distinguished: a first unentangled regime where the viscosity increase rate strongly depends on hydrophobic content and a second entangled regime where the viscosity follows a scaling behavior of the polymer concentration with an exponent close to 4. Introduction Water-soluble polymers are of great interest from the scientific as well as the technological point of view. These polymers have been subjected to extensive research [1]. Water-soluble polymers are present in the composition of different types of industrial formulations as stabilizing, flocculants, absorbent, emulsifiers, thickeners, etc. -

Prediction of Metabolic Stability and Bioavailability with Bioisosteric Replacements
Prediction of Metabolic Stability and Bioavailability with Bioisosteric Replacements Alison Pui Ki Choy Clare College University of Cambridge Date of submission: September 2017 This dissertation is submitted for the degree of Doctor of Philosophy. Prediction of Metabolic Stability and Bioavailability with Bioisosteric Replacements Alison Pui Ki Choy Abstract Drug development is a long and expensive process. Potential drug candidates can fail clinical trials due to numerous issues, including metabolic stability and efficacy issues, wasting years of research effort and resource. This thesis detailed the development of in silico methods to predict the metabolic stability of structures and their bioavailability. Coralie Atom-based Statistical SOM Identifier (CASSI) is a site of metabolism (SOM) predictor which provides a SOM prediction based on statistical information gathered about previously seen atoms present in similar environments. CASSI is a real-time SOM predictor accessible via graphical user interface (GUI), allowing users to view the prediction results and likelihood of each atom to undergo different types of metabolic transformation. Fast Metabolizer (FAME)1 is a ligand-based SOM predictor developed around the same time by Kirchmair et al. In the course of the evaluation of CASSI and FAME performance, the two concepts were combined to produce FamePrint. FamePrint is a tool developed within the Coralie Cheminformatics Platform developed by Lhasa Limited. which can carry out SOM predictions, as well as bioisosteric replacement identification. Same as CASSI, this is available via the Coralie application GUI. The bioavailability issues caused by the metabolic enzyme, cytochrome P450 3A4, and transporter protein P-gylcoprotein are also investigated in this work, along with the potential synergistic relationship between the two systems. -

Applications of Ionic Liquids in Membrane Separation
University of Arkansas, Fayetteville ScholarWorks@UARK Theses and Dissertations 12-2019 Applications of Ionic Liquids in Membrane Separation Mohanad Kamaz University of Arkansas, Fayetteville Follow this and additional works at: https://scholarworks.uark.edu/etd Part of the Complex Fluids Commons, Membrane Science Commons, and the Transport Phenomena Commons Citation Kamaz, M. (2019). Applications of Ionic Liquids in Membrane Separation. Theses and Dissertations Retrieved from https://scholarworks.uark.edu/etd/3491 This Dissertation is brought to you for free and open access by ScholarWorks@UARK. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of ScholarWorks@UARK. For more information, please contact [email protected]. Applications of Ionic Liquids in Membrane Separation A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Engineering by Mohanad Kamaz University of Basrah Bachelor of Science in Chemical Engineering, 2011 University of Arkansas Master of Science in Chemical Engineering, 2017 December 2019 University of Arkansas This dissertation is approved for recommendation to the Graduate Council. ____________________________________ Ranil Wickramasinghe, Ph.D. Dissertation Director ____________________________________ ____________________________________ David Ford, Ph.D. Ed Clauson, Ph.D. Committee Member Committee Member ____________________________________ ____________________________________ Xianghong Qian, Ph.D. Wen Zhang, Ph.D. Committee Member Committee Member Abstract Ionic liquids represent an emerging attractive material in membrane technology. The central theme of this doctoral dissertation is to develope novel membranes using ionic liquids. Two different approaches were used to prepare ionic liquid membranes including the immobilization of ionic liquid within the membrane pores or the use of pressure assembly method to deposit a selective ionic liquid layer on the top membrane surface. -

Nonionic Surfactant to Enhance the Performances of Alkaline–Surfactant–Polymer Flooding with a Low Salinity Constraint
applied sciences Article Nonionic Surfactant to Enhance the Performances of Alkaline–Surfactant–Polymer Flooding with a Low Salinity Constraint Shabrina Sri Riswati 1, Wisup Bae 1,*, Changhyup Park 2,* , Asep K. Permadi 3 and Adi Novriansyah 1 1 Department of Energy and Mineral Resources Engineering, Sejong University, Seoul 05006, Korea; [email protected] (S.S.R.); [email protected] (A.N.) 2 Department of Energy and Resources Engineering, Kangwon National University, Chuncheon, Kangwon 24341, Korea 3 Petroleum Engineering Department, Bandung Institute of Technology, Bandung 40132, Indonesia; [email protected] * Correspondence: [email protected] (W.B.); [email protected] (C.P.); Tel.: +82-33-2506259 (C.P.) Received: 12 May 2020; Accepted: 25 May 2020; Published: 28 May 2020 Abstract: This paper presents a nonionic surfactant in the anionic surfactant pair (ternary mixture) that influences the hydrophobicity of the alkaline–surfactant–polymer (ASP) slug within low-salinity formation water, an environment that constrains optimal designs of the salinity gradient and phase types. The hydrophobicity effectively reduced the optimum salinity, but achieving as much by mixing various surfactants has been challenging. We conducted a phase behavior test and a coreflooding test, and the results prove the effectiveness of the nonionic surfactant in enlarging the chemical applicability by making ASP flooding more hydrophobic. The proposed ASP mixture consisted of 0.2 wt% sodium carbonate, 0.25 wt% anionic surfactant pair, and 0.2 wt% nonionic surfactant, and 0.15 wt% hydrolyzed polyacrylamide. The nonionic surfactant decreased the optimum salinity to 1.1 wt% NaCl compared to the 1.7 wt% NaCl of the reference case with heavy alcohol present instead of the nonionic surfactant. -

Appendix C Medication Tables
Appendix C Medication Tables Note: The medication tables are not meant to be inclusive lists of all available therapeutic agents. Approved medication tables will be updated regularly. Discrepancies must be reported. See Resource Section of this manual for additional contact information. Release Notes: Aspirin Table Version 1.0 Table 1.1 Aspirin and Aspirin-Containing Medications Acetylsalicylic Acid Acuprin 81 Alka-Seltzer Alka-Seltzer Morning Relief Anacin Arthritis Foundation Aspirin Arthritis Pain Ascriptin Arthritis Pain Formula ASA ASA Baby ASA Baby Chewable ASA Baby Coated ASA Bayer ASA Bayer Children's ASA Buffered ASA Children's ASA EC ASA Enteric Coated ASA/Maalox Ascriptin Aspergum Aspir-10 Aspir-Low Aspir-Lox Aspir-Mox Aspir-Trin Aspirbuf Aspircaf Aspirin Aspirin Baby Aspirin Bayer Aspirin Bayer Children's Aspirin Buffered Aspirin Child Aspirin Child Chewable Aspirin Children's Aspirin EC Aspirin Enteric Coated Specifications Manual for National Appendix C-1 Hospital Quality Measures Table 1.1 Aspirin and Aspirin-Containing Medications (continued) Aspirin Litecoat Aspirin Lo-Dose Aspirin Low Strength Aspirin Tri-Buffered Aspirin, Extended Release Aspirin/butalbital/caffeine Aspirin/caffeine Aspirin/pravachol Aspirin/pravastatin Aspirtab Bayer Aspirin Bayer Aspirin PM Extra Strength Bayer Children’s Bayer EC Bayer Enteric Coated Bayer Low Strength Bayer Plus Buffered ASA Buffered Aspirin Buffered Baby ASA Bufferin Bufferin Arthritis Strength Bufferin Extra Strength Buffex Cama Arthritis Reliever Child’s Aspirin Coated Aspirin