Antigen Binding Protein and Its Use As Addressing Product for the Treatment of Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methods for Predicting the Survival Time of Patients Suffering from a Lung Cancer
THETWO TORTOITUUSN 20180252720A1ULLUM HOLATIN ( 19) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 / 0252720 A1 DIEU -NOSJEAN et al. (43 ) Pub. Date : Sep . 6 , 2018 (54 ) METHODS FOR PREDICTING THE Publication Classification SURVIVAL TIME OF PATIENTS SUFFERING (51 ) Int . Ci. FROM A LUNG CANCER GOIN 33 /574 ( 2006 .01 ) (71 ) Applicants : INSERM (INSTITUT NATIONAL ( 52 ) U . S . CI. DE LA SANTE ET DE LA CPC .. GOIN 33 /57423 (2013 . 01) ; GOIN 2800/ 52 RECHERCHE MEDICALE ) , Paris ( 2013 . 01 ) (FR ) ; UNIVERSITE PARIS DESCARTES , Paris ( FR ) ; SORBONNE UNIVERSITE , Paris (57 ) ABSTRACT (FR ) ; UNIVERSITE PARIS DIDEROT - PARIS 7 , Paris ( FR ) ; The present invention relates to methods for predicting the ASSISTANCE survival time of patients suffering from a lung cancer . In PUBLIQUE -HOPITAUX DE PARIS particular , the present invention relates to a method for (ADHP ) , Paris (FR ) predicting the survival time of a subject suffering from a lung cancer comprising the steps of i) quantifying the ( 72 ) Inventors: Marie - Caroline DIEU - NOSJEAN , density of regulatory T ( Treg ) cells in a tumor tissue sample Paris (FR ) ; Wolf Herdman obtained from the subject, ii ) quantifying the density of one FRIDMAN , Paris ( FR ) ; Catherine further population of immune cells selected from the group SAUTES - FRIDMAN , Paris ( FR ) ; consisting of TLS -mature DC or TLS - B cells or Tconv cells , Priyanka DEVI, Paris (FR ) CD8 + T cells or CD8 + Granzyme - B + T cells in said tumor tissue sample , iii ) comparing the densities quantified at steps (21 ) Appl. No. : 15/ 754 , 640 i ) and ii ) with their corresponding predetermined reference values and iv ) concluding that the subject will have a short ( 22 ) PCT Filed : Aug . -
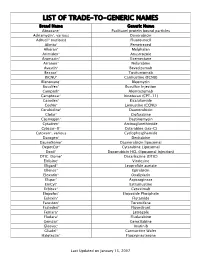
Trade-To-Generic Names
LIST OF TRADE-TO-GENERIC NAMES Brand Name Generic Name Abraxane® Paclitaxel protein bound particles Adriamycin®, various Doxorubicin Adrucil® (various) Fluorouracil Alimta® Pemetrexed Alkeran® Melphalan Arimidex® Anastrozole Aromasin® Exemestane Arranon® Nelarabine Avastin® Bevacizumab Bexxar® Tositumomab BiCNU® Carmustine (BCNU) Blenoxane® Bleomycin Busulfex® Busulfan Injection Campath® Alemtuzumab Camptosar® Irinotecan (CPT-11) Casodex® Bicalutamide CeeNu® Lomustine (CCNU) Cerubidine® Daunorubicin Clolar® Clofarabine Cosmegen® Dactinomycin Cytadren® Aminoglutethimide Cytosar-U® Cytarabine (ara-C) Cytoxan®, various Cyclophosphamide Dacogen® Decitabine DaunoXome® Daunorubicin liposomal DepotCyt® Cytarabine Liposomal Doxil® Doxorubicin HCL (liposomal injection) DTIC-Dome® Dacarbazine (DTIC) Eldisine® Vindesine Eligard® Leuprolide acetate Ellence® Epirubicin Eloxatin® Oxaliplatin Elspar® Asparaginase EmCyt® Estramustine Erbitux® Cetuximab Etopofos® Etoposide Phosphate Eulexin® Flutamide Fareston® Toremifene Faslodex® Fluvestrant Femara® Letrozole Fludara® Fludarabine Gemzar® Gemcitabine Gleevec® Imatinib Gliadel® Carmustine Wafer Halotestin® Fluoxymesterone Last Updated on January 15, 2007 Brand Name Generic Name Herceptin® Trastuzumab Hexalen® Altretamine Hycamtin® Topotecan Hydrea® Hydroxyurea Idamycin® Idarubicin Ifex® Ifosfamide Intron A® Interferon alfa-2b Iressa® Gefitinib Leukeran® Chlorambucil Leukine® Sargramostim Leustatin® Cladribine Lupron depot® Leuprolide acetate depot Lupron® Leuprolide acetate Matulane® Procarbazine Megace® -

Table of Contents
ANTICANCER RESEARCH International Journal of Cancer Research and Treatment ISSN: 0250-7005 Volume 32, Number 4, April 2012 Contents Experimental Studies * Review: Multiple Associations Between a Broad Spectrum of Autoimmune Diseases, Chronic Inflammatory Diseases and Cancer. A.L. FRANKS, J.E. SLANSKY (Aurora, CO, USA)............................................ 1119 Varicella Zoster Virus Infection of Malignant Glioma Cell Cultures: A New Candidate for Oncolytic Virotherapy? H. LESKE, R. HAASE, F. RESTLE, C. SCHICHOR, V. ALBRECHT, M.G. VIZOSO PINTO, J.C. TONN, A. BAIKER, N. THON (Munich; Oberschleissheim, Germany; Zurich, Switzerland) .................................... 1137 Correlation between Adenovirus-neutralizing Antibody Titer and Adenovirus Vector-mediated Transduction Efficiency Following Intratumoral Injection. K. TOMITA, F. SAKURAI, M. TACHIBANA, H. MIZUGUCHI (Osaka, Japan) .......................................................................................................... 1145 Reduction of Tumorigenicity by Placental Extracts. A.M. MARLEAU, G. MCDONALD, J. KOROPATNICK, C.-S. CHEN, D. KOOS (Huntington Beach; Santa Barbara; Loma Linda; San Diego, CA, USA; London, ON, Canada) ...................................................................................................................................... 1153 Stem Cell Markers as Predictors of Oral Cancer Invasion. A. SIU, C. LEE, D. DANG, C. LEE, D.M. RAMOS (San Francisco, CA, USA) ................................................................................................ -

Journal of Pharmacology and Experimental Therapeutics
Journal of Pharmacology and Experimental Therapeutics Molecular Determinants of Ligand Selectivity for the Human Multidrug And Toxin Extrusion Proteins, MATE1 and MATE-2K Bethzaida Astorga, Sean Ekins, Mark Morales and Stephen H Wright Department of Physiology, University of Arizona, Tucson, AZ 85724, USA (B.A., M.M., and S.H.W.) Collaborations in Chemistry, 5616 Hilltop Needmore Road, Fuquay-Varina NC 27526, USA (S.E.) Supplemental Table 1. Compounds selected by the common features pharmacophore after searching a database of 2690 FDA approved compounds (www.collaborativedrug.com). FitValue Common Name Indication 3.93897 PYRIMETHAMINE Antimalarial 3.3167 naloxone Antidote Naloxone Hydrochloride 3.27622 DEXMEDETOMIDINE Anxiolytic 3.2407 Chlordantoin Antifungal 3.1776 NALORPHINE Antidote Nalorphine Hydrochloride 3.15108 Perfosfamide Antineoplastic 3.11759 Cinchonidine Sulfate Antimalarial Cinchonidine 3.10352 Cinchonine Sulfate Antimalarial Cinchonine 3.07469 METHOHEXITAL Anesthetic 3.06799 PROGUANIL Antimalarial PROGUANIL HYDROCHLORIDE 100MG 3.05018 TOPIRAMATE Anticonvulsant 3.04366 MIDODRINE Antihypotensive Midodrine Hydrochloride 2.98558 Chlorbetamide Antiamebic 2.98463 TRIMETHOPRIM Antibiotic Antibacterial 2.98457 ZILEUTON Antiinflammatory 2.94205 AMINOMETRADINE Diuretic 2.89284 SCOPOLAMINE Antispasmodic ScopolamineHydrobromide 2.88791 ARTICAINE Anesthetic 2.84534 RITODRINE Tocolytic 2.82357 MITOBRONITOL Antineoplastic Mitolactol 2.81033 LORAZEPAM Anxiolytic 2.74943 ETHOHEXADIOL Insecticide 2.64902 METHOXAMINE Antihypotensive Methoxamine -

Lonidamine Induces Apoptosis in Drug-Resistant Cells Independently of the P53 Gene
Lonidamine induces apoptosis in drug-resistant cells independently of the p53 gene. D Del Bufalo, … , A Sacchi, G Zupi J Clin Invest. 1996;98(5):1165-1173. https://doi.org/10.1172/JCI118900. Research Article Lonidamine, a dichlorinated derivative of indazole-3-carboxylic acid, was shown to play a significant role in reversing or overcoming multidrug resistance. Here, we show that exposure to 50 microg/ml of lonidamine induces apoptosis in adriamycin and nitrosourea-resistant cells (MCF-7 ADR(r) human breast cancer cell line, and LB9 glioblastoma multiform cell line), as demonstrated by sub-G1 peaks in DNA content histograms, condensation of nuclear chromatin, and internucleosomal DNA fragmentation. Moreover, we find that apoptosis is preceded by accumulation of the cells in the G0/G1 phase of the cell cycle. Interestingly, lonidamine fails to activate the apoptotic program in the corresponding sensitive parental cell lines (ADR-sensitive MCF-7 WT, and nitrosourea-sensitive LI cells) even after long exposure times. The evaluation of bcl-2 protein expression suggests that this different effect of lonidamine treatment in drug-resistant and -sensitive cell lines might not simply be due to dissimilar expression levels of bcl-2 protein. To determine whether the lonidamine-induced apoptosis is mediated by p53 protein, we used cells lacking endogenous p53 and overexpressing either wild-type p53 or dominant-negative p53 mutant. We find that apoptosis by lonidamine is independent of the p53 gene. Find the latest version: https://jci.me/118900/pdf -

Effect of Lonidamine on Systemic Therapy of DB-1 Human Melanoma Xenografts with Temozolomide KAVINDRA NATH 1, DAVID S
ANTICANCER RESEARCH 37 : 3413-3421 (2017) doi:10.21873/anticanres.11708 Effect of Lonidamine on Systemic Therapy of DB-1 Human Melanoma Xenografts with Temozolomide KAVINDRA NATH 1, DAVID S. NELSON 1, JEFFREY ROMAN 1, MARY E. PUTT 2, SEUNG-CHEOL LEE 1, DENNIS B. LEEPER 3 and JERRY D. GLICKSON 1 Departments of 1Radiology and 2Biostatistics & Epidemiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, U.S.A.; 3Department of Radiation Oncology, Thomas Jefferson University, Philadelphia, PA, U.S.A. Abstract. Background/Aim: Since temozolomide (TMZ) is stages. However, following recurrence with metastasis, the activated under alkaline conditions, we expected lonidamine prognosis is poor. Mutationally-activated BRAF is found in (LND) to have no effect or perhaps diminish its activity, but 40-60% of all melanomas with most common substitution of initial results suggest it may actually enhance either or both valine to glutamic acid at codon 600 (p. V600E) (3). Overall short- and long-term activity of TMZ in melanoma xenografts. survival is approaching two years using agents that target this Materials and Methods: Cohorts of 5 mice with subcutaneous mutation (4, 5). MEK, RAS and other signal transduction xenografts ~5 mm in diameter were treated with saline inhibitors, in combination with mutant BRAF inhibitors, have (control (CTRL)), LND only, TMZ only or LND followed by been used to deal with melanoma resistance to these agents TMZ at t=40 min (time required for maximal tumor (6). Treatment with anti-programmed death-1 (PD-1) acidification). Results: Mean tumor volume for LND+TMZ for checkpoint inhibitor immunotherapy currently produces the period between 6 and 26 days was reduced compared to durable response in about 25% of melanoma patients (7-10). -

Mepact, INN-Mifamurtide
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT MEPACT 4 mg powder for concentrate for dispersion for infusion 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each vial contains 4 mg mifamurtide*. After reconstitution, each mL of suspension in the vial contains 0.08 mg mifamurtide. *fully synthetic analogue of a component of Mycobacterium sp. cell wall. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder for concentrate for dispersion for infusion White to off-white homogeneous cake or powder. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications MEPACT is indicated in children, adolescents and young adults for the treatment of high-grade resectable non-metastatic osteosarcoma after macroscopically complete surgical resection. It is used in combination with post-operative multi-agent chemotherapy. Safety and efficacy have been assessed in studies of patients 2 to 30 years of age at initial diagnosis (see section 5.1). 4.2 Posology and method of administration Mifamurtide treatment should be initiated and supervised by specialist physicians experienced in the diagnosis and treatment of osteosarcoma. Posology The recommended dose of mifamurtide for all patients is 2 mg/m2 body surface area. It should be administered as adjuvant therapy following resection: twice weekly at least 3 days apart for 12 weeks, followed by once-weekly treatments for an additional 24 weeks for a total of 48 infusions in 36 weeks. Special populations Adults > 30 years None of the patients treated in the osteosarcoma studies were 65 years or older and in the phase III randomised study, only patients up to the age of 30 years were included. -

Advances and Limitations of Antibody Drug Conjugates for Cancer
biomedicines Review Advances and Limitations of Antibody Drug Conjugates for Cancer Candice Maria Mckertish and Veysel Kayser * Sydney School of Pharmacy, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia; [email protected] * Correspondence: [email protected]; Tel.: +61-2-9351-3391 Abstract: The popularity of antibody drug conjugates (ADCs) has increased in recent years, mainly due to their unrivalled efficacy and specificity over chemotherapy agents. The success of the ADC is partly based on the stability and successful cleavage of selective linkers for the delivery of the payload. The current research focuses on overcoming intrinsic shortcomings that impact the successful devel- opment of ADCs. This review summarizes marketed and recently approved ADCs, compares the features of various linker designs and payloads commonly used for ADC conjugation, and outlines cancer specific ADCs that are currently in late-stage clinical trials for the treatment of cancer. In addition, it addresses the issues surrounding drug resistance and strategies to overcome resistance, the impact of a narrow therapeutic index on treatment outcomes, the impact of drug–antibody ratio (DAR) and hydrophobicity on ADC clearance and protein aggregation. Keywords: antibody drug conjugates; drug resistance; linkers; payloads; therapeutic index; target specific; ADC clearance; protein aggregation Citation: Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for 1. Introduction Cancer. Biomedicines 2021, 9, 872. Conventional cancer therapy often entails a low therapeutic window and non-specificity https://doi.org/10.3390/ of chemotherapeutic agents that consequently affects normal cells with high mitotic rates biomedicines9080872 and provokes an array of adverse effects, and in some cases leads to drug resistance [1]. -

Modifications to the Harmonized Tariff Schedule of the United States to Implement Changes to the Pharmaceutical Appendix
United States International Trade Commission Modifications to the Harmonized Tariff Schedule of the United States to Implement Changes to the Pharmaceutical Appendix USITC Publication 4208 December 2010 U.S. International Trade Commission COMMISSIONERS Deanna Tanner Okun, Chairman Irving A. Williamson, Vice Chairman Charlotte R. Lane Daniel R. Pearson Shara L. Aranoff Dean A. Pinkert Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Modifications to the Harmonized Tariff Schedule of the United States to Implement Changes to the Pharmaceutical Appendix Publication 4208 December 2010 (This page is intentionally blank) Pursuant to the letter of request from the United States Trade Representative of December 15, 2010, set forth at the end of this publication, and pursuant to section 1207(a) of the Omnibus Trade and Competitiveness Act, the United States International Trade Commission is publishing the following modifications to the Harmonized Tariff Schedule of the United States (HTS) to implement changes to the Pharmaceutical Appendix, effective on January 1, 2011. Table 1 International Nonproprietary Name (INN) products proposed for addition to the Pharmaceutical Appendix to the Harmonized Tariff Schedule INN CAS Number Abagovomab 792921-10-9 Aclidinium Bromide 320345-99-1 Aderbasib 791828-58-5 Adipiplon 840486-93-3 Adoprazine 222551-17-9 Afimoxifene 68392-35-8 Aflibercept 862111-32-8 Agatolimod -

Phenotype Microarrays Panels PM-M1 to PM-M14
Phenotype MicroArrays™ Panels PM-M1 to PM-M14 for Phenotypic Characterization of Mammalian Cells Assays: Energy Metabolism Pathways Ion and Hormone Effects on Cells Sensitivity to Anti-Cancer Agents and for Optimizing Culture Conditions for Mammalian Cells PRODUCT DESCRIPTIONS AND INSTRUCTIONS FOR USE PM-M1 Cat. #13101 PM-M2 Cat. #13102 PM-M3 Cat. #13103 PM-M4 Cat. #13104 PM-M5 Cat. #13105 PM-M6 Cat. #13106 PM-M7 Cat. #13107 PM-M8 Cat. #13108 PM-M11 Cat. #13111 PM-M12 Cat. #13112 PM-M13 Cat. #13113 PM-M14 Cat. #13114 © 2016 Biolog, Inc. All rights reserved Printed in the United States of America 00P 134 Rev F February 2020 - 1 - CONTENTS I. Introduction ...................................................................................................... 2 a. Overview ................................................................................................... 2 b. Background ............................................................................................... 2 c. Uses ........................................................................................................... 2 d. Advantages ................................................................................................ 3 II. Product Description, PM-M1 to M4 ................................................................ 3 III. Protocols, PM-M1 to M4 ................................................................................. 7 a. Materials Required .................................................................................... 7 b. Determination -

Chemical Properties Biological Description Solubility
Data Sheet (Cat.No.T4412) Roquinimex Chemical Properties CAS No.: 84088-42-6 Formula: C18H16N2O3 Molecular Weight: 308.34 Appearance: N/A Storage: 0-4℃ for short term (days to weeks), or -20℃ for long term (months). Biological Description Description Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity; inhibits angiogenesis and reduces the secretion of TNF alpha. Targets(IC50) Others: None In vivo Prophylactic administration of DSS-treated mice with roquinimex significantly reduced clinical signs of colitis, MDS and the CH-reduction. Moreover, in roquinimex treated animals, the MPO activity was significantly reduced by more than 50% compared to DSS control mice. Notably, therapeutic administration of roquinimex in DSS-treated mice also significantly inhibited the MDS, CH-reduction and MPO activity. Linomide, a synthetic immunomodulator, at concentrations effective in vivo reduces the number of MBP-reactive TNF-alpha and increases MBP-reactive IL-10 and TGF-beta mRNA expressing MNC from MS patients' blood when analysed in vitro. Compared to dexamethasone, Linomide up-regulated levels of blood MNC expressing mRNA of TGF-beta after culture in presence of MBP Solubility Information Solubility DMSO: 83.3 mg/mL (270.17 mM) (< 1 mg/ml refers to the product slightly soluble or insoluble) Preparing Stock Solutions 1mg 5mg 10mg 1 mM 3.243 mL 16.216 mL 32.432 mL 5 mM 0.649 mL 3.243 mL 6.486 mL 10 mM 0.324 mL 1.622 mL 3.243 mL 50 mM 0.065 mL 0.324 mL 0.649 mL Please select the appropriate solvent to prepare the stock solution, according to the solubility of the product in different solvents. -

Encapsulation of Nedaplatin in Novel Pegylated Liposomes Increases Its Cytotoxicity and Genotoxicity Against A549 and U2OS Human Cancer Cells
pharmaceutics Article Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells 1, 2, 1 1 2, Salma El-Shafie y, Sherif Ashraf Fahmy y , Laila Ziko , Nada Elzahed , Tamer Shoeib * and Andreas Kakarougkas 1,* 1 Department of Biology, School of Sciences and Engineering, The American University in Cairo, Cairo 11835, Egypt; [email protected] (S.E.-S.); [email protected] (L.Z.); [email protected] (N.E.) 2 Department of Chemistry, School of Sciences and Engineering, The American University in Cairo, Cairo 11835 Egypt; sheriff[email protected] * Correspondence: [email protected] (T.S.); [email protected] (A.K.) These authors contribute equally to this paper. y Received: 7 April 2020; Accepted: 25 August 2020; Published: 10 September 2020 Abstract: Following the discovery of cisplatin over 50 years ago, platinum-based drugs have been a widely used and effective form of cancer therapy, primarily causing cell death by inducing DNA damage and triggering apoptosis. However, the dose-limiting toxicity of these drugs has led to the development of second and third generation platinum-based drugs that maintain the cytotoxicity of cisplatin but have a more acceptable side-effect profile. In addition to the creation of new analogs, tumor delivery systems such as liposome encapsulated platinum drugs have been developed and are currently in clinical trials. In this study, we have created the first PEGylated liposomal form of nedaplatin using thin film hydration. Nedaplatin, the main focus of this study, has been exclusively used in Japan for the treatment of non-small cell lung cancer, head and neck, esophageal, bladder, ovarian and cervical cancer.