Genetic and Flow Anomalies in Congenital Heart Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fetal Blood Flow and Genetic Mutations in Conotruncal Congenital Heart Disease
Journal of Cardiovascular Development and Disease Review Fetal Blood Flow and Genetic Mutations in Conotruncal Congenital Heart Disease Laura A. Dyer 1 and Sandra Rugonyi 2,* 1 Department of Biology, University of Portland, Portland, OR 97203, USA; [email protected] 2 Department of Biomedical Engineering, Oregon Health & Science University, Portland, OR 97239, USA * Correspondence: [email protected] Abstract: In congenital heart disease, the presence of structural defects affects blood flow in the heart and circulation. However, because the fetal circulation bypasses the lungs, fetuses with cyanotic heart defects can survive in utero but need prompt intervention to survive after birth. Tetralogy of Fallot and persistent truncus arteriosus are two of the most significant conotruncal heart defects. In both defects, blood access to the lungs is restricted or non-existent, and babies with these critical conditions need intervention right after birth. While there are known genetic mutations that lead to these critical heart defects, early perturbations in blood flow can independently lead to critical heart defects. In this paper, we start by comparing the fetal circulation with the neonatal and adult circulation, and reviewing how altered fetal blood flow can be used as a diagnostic tool to plan interventions. We then look at known factors that lead to tetralogy of Fallot and persistent truncus arteriosus: namely early perturbations in blood flow and mutations within VEGF-related pathways. The interplay between physical and genetic factors means that any one alteration can cause significant disruptions during development and underscore our need to better understand the effects of both blood flow and flow-responsive genes. -

The Role of Echocardiography in the Management of Adult Patients with Congenital Heart Disease Following Operative Treatment
779 Review Article The role of echocardiography in the management of adult patients with congenital heart disease following operative treatment Kálmán Havasi, Nóra Ambrus, Anita Kalapos, Tamás Forster, Attila Nemes 2nd Department of Medicine and Cardiology Centre, Medical Faculty, Albert Szent-Györgyi Clinical Centre, University of Szeged, Szeged, Hungary Contributions: (I) Conception and design: K Havasi; (II) Administrative support: N Ambrus, A Kalapos; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: K Havasi; (V) Data analysis and interpretation: K Havasi, N Ambrus, A Kalapos; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Attila Nemes, MD, PhD, DSc, FESC. 2nd Department of Medicine and Cardiology Centre, Medical Faculty, Albert Szent-Györgyi Clinical Centre, University of Szeged, H-6725 Szeged, Semmelweis street 8, Szeged, Hungary. Email: [email protected]. Abstract: Treatment of congenital heart diseases has significantly advanced over the last few decades. Due to the continuously increasing survival rate, there are more and more adult patients with congenital heart diseases and these patients present at the adult cardiologist from the paediatric cardiology care. The aim of the present review is to demonstrate the role of echocardiography in some significant congenital heart diseases. Keywords: Echocardiography; congenital heart disease; adult Submitted Jul 03, 2018. Accepted for publication Sep 07, 2018. doi: 10.21037/cdt.2018.09.11 View this article at: http://dx.doi.org/10.21037/cdt.2018.09.11 Introduction if the patient’s condition is evaluated by the proper imaging modality. Therefore, the treating physician should know the Treatment of congenital heart diseases has significantly benefits, disadvantages and clinical indications of the certain advanced during the last few decades. -

Prenatal Development Timeline
Prenatal Development Timeline Nervous Cardiovascular Muscular Early Events Special Senses Respiratory Skeletal Growth Parameters Blood & Immune Gastrointestinal Endocrine General Skin/Integument Renal/Urinary Reproductive Movement Unit 1: The First Week Day 0 — — Embryonic period begins Fertilization resulting in zygote formation Day 1 — — Embryo is spherically shaped with 12 to 16 cells Day 1 - Day 1 — — Fertilization - development begins with a single-cell embryo!!! Day 2 — — Early pregnancy factor (EPF) Activation of the genome Zygote divides into two blastomeres (24 – 30 hours from start of fertilization) Day 4 — — Embryonic disc Free floating blastocyst Hypoblast & epiblast Inner cell mass See where the back and chest will be Day 5 — — Hatching blastocyst Day 6 — — Embryo attaches to wall of uterus 1 week — — Chorion Placenta begins to form Unit 2: 1 to 2 Weeks 1 week, 1 day — — Amnioblasts present; amnion and amniotic cavity formation begins Positive pregnancy test 1 week, 2 days — — Cells in womb engorged with nutrients 1 week, 4 days — — Longitudinal axis 1 week, 5 days — — Implantation complete Yolk sac 1 week, 6 days — — Primordial blood vessels Amnion with single cell layer Chorionic villi 2 weeks — — Yolk sac Yolk sac Unit 3: 2 to 3 Weeks 2 weeks, 1 day — — 3 germ layers Rostral-caudal orientation 2 weeks, 2 days — — Erythroblasts in yolk sac Three types of blood-forming cells in yolk sac Amnion with two cell layers Secondary villi www.ehd.org 1 of 10 2 weeks, 4 days — — Foregut, midgut, and hindgut Brain is first organ to -

Tetralogy of Fallot Clinical Research Study
Tetralogy of Fallot Clinical Research Study • Research Goal: Test a new way to look at the right side of the heart by new ECHO technology and compare that with MRI • The stu dy v is it w ill last approx imate ly 3 h ours an d ilinvolves a cardiac MRI and ECHO • 9 years old and up with Tetralogy of Fallot • No devices such as ICD or Pacemaker • No RV to PA conduit • For more information, contact T. Aaron West at [email protected] or call (614)355-3448 • Research incentive offered ………………..…………………………………………………………………………………………………………………………………….. Tetralogy of Fallot Past, Present, Future Stephen R. Crumb, APN Coordinator, COACH Program Columbus Ohio Adult Congenital Heart ………………..…………………………………………………………………………………………………………………………………….. Disclosure: Go Buck(y)(s) ? Survival to 18 years of age with CHD 1980 – 90 1970 – 75 Year Born with CHD 1960 – 40 1940 – 20 0 10 20 30 40 50 60 70 80 90 100 ………………..…………………………………………………………………………………………………………………………………….. Pediatric to Adult Congenital Heart Disease Expanding Population of Adol escents and AdAdlults Increased Mid Term with CHD Survival Increased Early Lower Perioperative Survival Mortality Early Complete Improved Surgical Repair Techniques Advances in Fetal Diagnosis NICU Care Incidence of CHD ………………..…………………………………………………………………………………………………………………………………….. Ratio of Pediatric to Adult Patients with CHD Pediatric patients Adult patients 1965 1985 2005 ………………..…………………………………………………………………………………………………………………………………….. Normal Heart Tetralogy of Fallot ETIENNE-LOUIS ARTHUR FALLOT Mixing red and blue -

Massachusetts Birth Defects 2002-2003
Massachusetts Birth Defects 2002-2003 Massachusetts Birth Defects Monitoring Program Bureau of Family Health and Nutrition Massachusetts Department of Public Health January 2008 Massachusetts Birth Defects 2002-2003 Deval L. Patrick, Governor Timothy P. Murray, Lieutenant Governor JudyAnn Bigby, MD, Secretary, Executive Office of Health and Human Services John Auerbach, Commissioner, Massachusetts Department of Public Health Sally Fogerty, Director, Bureau of Family Health and Nutrition Marlene Anderka, Director, Massachusetts Center for Birth Defects Research and Prevention Linda Casey, Administrative Director, Massachusetts Center for Birth Defects Research and Prevention Cathleen Higgins, Birth Defects Surveillance Coordinator Massachusetts Department of Public Health 617-624-5510 January 2008 Acknowledgements This report was prepared by the staff of the Massachusetts Center for Birth Defects Research and Prevention (MCBDRP) including: Marlene Anderka, Linda Baptiste, Elizabeth Bingay, Joe Burgio, Linda Casey, Xiangmei Gu, Cathleen Higgins, Angela Lin, Rebecca Lovering, and Na Wang. Data in this report have been collected through the efforts of the field staff of the MCBDRP including: Roberta Aucoin, Dorothy Cichonski, Daniel Sexton, Marie-Noel Westgate and Susan Winship. We would like to acknowledge the following individuals for their time and commitment to supporting our efforts in improving the MCBDRP. Lewis Holmes, MD, Massachusetts General Hospital Carol Louik, ScD, Slone Epidemiology Center, Boston University Allen Mitchell, -

Endocardial Cushion and Myocardial Defects After Cardiac Myocyte-Specific Conditional Deletion of the Bone Morphogenetic Protein Receptor ALK3
Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3 Vinciane Gaussin*†, Tom Van de Putte‡, Yuji Mishina§, Mark C. Hanks¶, An Zwijsen‡, Danny Huylebroeck‡, Richard R. Behringerʈ, and Michael D. Schneider*,** *Center for Cardiovascular Development, Baylor College of Medicine, Houston, TX 77030; ‡Flanders Interuniversity Institute for Biotechnology (VIB07), K.U. Leuven, 3000 Leuven, Belgium; §National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709; ¶Procter and Gamble Pharmaceuticals Health Care Research Center, 8700 Mason Montgomery Road, Mason, OH 45040; and ʈUniversity of Texas–M. D. Anderson Cancer Center, Houston, TX 77030 Edited by Eric N. Olson, University of Texas Southwestern Medical Center, Dallas, TX, and approved December 31, 2001 (received for review July 26, 2001) Receptors for bone morphogenetic proteins (BMPs), members of velopment, whereas ALK6 is absent from the heart at mid- the transforming growth factor- (TGF) superfamily, are persis- gestation (17). The developing heart also expresses ALK2͞ tently expressed during cardiac development, yet mice lacking type ActRIA (5, 18), which can function as a type I BMP receptor II or type IA BMP receptors die at gastrulation and cannot be used with preference for BMP6 and -7 (19). ALK3, ALK2, and to assess potential later roles in creation of the heart. Here, we BMPR-II are each essential for gastrulation and mesoderm used a Cre͞lox system for cardiac myocyte-specific deletion of the formation (18, 20, 21); mice lacking just BMP4 also fail to type IA BMP receptor, ALK3. ALK3 was specifically required at progress, typically, beyond the egg cylinder stage (22). -

Cardiovascular System Heart Development Cardiovascular System Heart Development
Cardiovascular System Heart Development Cardiovascular System Heart Development In human embryos, the heart begins to beat at approximately 22-23 days, with blood flow beginning in the 4th week. The heart is one of the earliest differentiating and functioning organs. • This emphasizes the critical nature of the heart in distributing blood through the vessels and the vital exchange of nutrients, oxygen, and wastes between the developing baby and the mother. • Therefore, the first system that completes its development in the embryo is called cardiovascular system. https://www.slideshare.net/DrSherifFahmy/intraembryonic-mesoderm-general-embryology Mesoderm is one of the three • Connective tissue primary germ layers that • Smooth and striated muscle • Cardiovascular System differentiates early in • Kidneys development that collectively • Spleen • Genital organs, ducts gives rise to all subsequent • Adrenal gland cortex tissues and organs. The cardiovascular system begins to develop in the third week of gestation. Blood islands develop in the newly formed mesoderm, and consist of (a) a central group of haemoblasts, the embryonic precursors of blood cells; (b) endothelial cells. Development of the heart and vascular system is often described together as the cardiovascular system. Development begins very early in mesoderm both within (embryonic) and outside (extra embryonic, vitelline, umblical and placental) the embryo. Vascular development occurs in many places. • Blood islands coalesce to form a vascular plexus. Preferential channels form arteries and veins. • Day 17 - Blood islands form first in the extra-embryonic mesoderm • Day 18 - Blood islands form next in the intra-embryonic mesoderm • Day 19 - Blood islands form in the cardiogenic mesoderm and coalesce to form a pair of endothelial heart tubes Development of a circulation • A circulation is established during the 4th week after the myocardium is differentiated. -
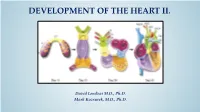
Development of Right Ventricle
DEVELOPMENT OF THE HEART II. David Lendvai M.D., Ph.D. Mark Kozsurek, M.D., Ph.D. • Septation of the common atrioventricular (AV) orifice. • Formation of the interatrial septum. • Formation of the muscular interventricular septum. • Appearance of the membranous interventricular septum and the spiral aorticopulmonary septum. right left septum primum septum primum septum primum septum primum septum primum septum primum foramen primum foramen primum septum primum septum primum foramen primum foramen primum septum primum septum primum foramen secundum foramen secundum foramen primum foramen primum septum primum foramen secundum septum primum foramen secundum foramen primum foramen primum septum primum septum primum foramen secundum foramen secundum septum secundum septum secundum foramen secundum foramen ovale foramen ovale septum primum septum primum septum secundum septum secundum foramen secundum foramen ovale foramen ovale septum primum septum primum septum secundum septum secundum foramen secundum septum primum foramen ovale foramen ovale septum primum SUMMARY • The septation of the common atrium starts with the appearance of the crescent-shaped septum primum. The opening of this septum, the foramen primum, becomes progressively smaller. • Before the foramen primum completly closes, postero-superiorly several small openings appear on the septum primum. These perforations coalesce later and form the foramen secundum. • On the right side of the septum primum a new septum, the septum secundum, starts to grow. The orifice of the septum secundum is the foramen ovale. • Finally two crescent-like, incomplete, partially overlapping septa exist with one hole on each. Septum secundum is more rigid and the septum primum on its left side acts as a valve letting the blood flow exclusively from the right to the left. -

Cardiovascular System Note: the Cardiovascular System Develops Early (Week 3), Enabling the Embryo to Grow Beyond the Short
Lymphatics: Lymph vessel formation is similar to blood angiogenesis. Lymphatics begin as lymph sacs in three regions: jugular (near brachiocephalic veins); cranial abdominal (future cysterna chyla); and iliac region. Lym- phatic vessels (ducts) form as outgrowths of the sacs. mesenchyme Lymph nodes are produced by localized mesoder- sinusoid lymph duct lumen mal invaginations that partition the vessel lumen into sinu- soids. The mesoderm develops a reticular framework within which mesodermal lymphocytes accumulate. The spleen and hemal nodes (in ruminants) invagination develop similar to the way lymph nodes develop. Lymph Node Formation Prior to birth, fetal circulation is designed for an in utero aqueous environment where the pla- centa oxygenates fetal blood. Suddenly, at birth... Three In-Utero Adjustments ductus Stretching and constriction of arteriosus umbilical arteries shifts fetal blood flow aortic arch from the placenta to the fetus. Reduced pulmonary trunk L atrium venous return through the (left) umbili- foramen ovale R cal vein and ductus venosus allows the atrium latter to gradually close (over a period caudal vena cava of days). Bradykinin released by expand- ductus venosus ing lungs and increased oxygen concen- tration in blood triggers constriction of aorta the ductus arteriosus which, over two liver months, is gradually converted to a fibrous structure, the ligamentum arte- umbilical v. riosum. portal v. The increased blood flow to the lungs and then to the left atrium equalizes pres- sure in the two atria, resulting in closure umbilical aa. of the foramen ovale that eventually grows permanent. 29 The cardiogenic area, the place where the embryonic heart originates, is located . -
Development and Teratology of Cardiovascular and Lymphatic Systems
Development and teratology of cardiovascular and lymphatic systems Repetition: Muscle tissue Beginning of the cardiovascular system development – the 3rd week: Hemangiogenesis (day 15 – 16) – blood islets (insulae sanguinae) in extraembryonic mesoderm and splanchnic mesenchyme of embryo Clusters of mesenchyme cells (angiogenic cells) differentiate into: - angioblasts endothelium (at the periphery of blood islets) - hemoblasts primitive erythrocytes (in the center of blood islets) Clusters of angiogenic cells form a "horseshoe-shaped" space between somatic and splanchnic layer of mesoderm = pericardial cavity. Two endothelial tubes arrise in splanchnic mesoderm. The ventral portion of these tubes forms the cardiogenic area with two heart tubes, while the lateral portions form the dorsal aortae. Germ disc: prosencephalon mesencephalon eye rhombencephalon heart lateral mesoderm somites small blood vessels blood islands 8,9 Spine primitive streak Initially, the cardiogenic area is located anterior to the prechordal plate and the neural plate. The growth of the central nervous system pulls the cardiogenic area and prechordal plate (buccopharyngeal membrane ventrally and caudally ( ). Cardiogenic region just cranial to the prechordal plate. The canalization of cardiogenic clusters in the splanchnic mesoderm results in the formation of the paired heart tubes. Folding of embryo and primitive gut separation from yolk sac. Fusion of the heart tubes a single heart tube is, temporarily attached to the dorsal side of the pericardial cavity by the -

Pretest Anatomy, Histology & Cell Biology
Anatomy, Histology, and Cell Biology PreTestTMSelf-Assessment and Review Notice Medicine is an ever-changing science. As new research and clinical experience broaden our knowledge, changes in treatment and drug therapy are required. The editors and the publisher of this work have checked with sources believed to be reli- able in their efforts to provide information that is complete and generally in accord with the standards accepted at the time of publication. However, in view of the pos- sibility of human error or changes in medical sciences, neither the editors nor the publisher nor any other party who has been involved in the preparation or publi- cation of this work warrants that the information contained herein is in every respect accurate or complete, and they are not responsible for any errors or omis- sions or for the results obtained from use of such information. Readers are encour- aged to confirm the information contained herein with other sources. For example and in particular, readers are advised to check the product information sheet included in the package of each drug they plan to administer to be certain that the information contained in this book is accurate and that changes have not been made in the recommended dose or in the contraindications for administration. This recommendation is of particular importance in connection with new or infre- quently used drugs. Anatomy, Histology, and Cell Biology PreTestTMSelf-Assessment and Review Third Edition Robert M. Klein, PhD Professor and Associate Dean Professional Development and Faculty Affairs Department of Anatomy and Cell Biology University of Kansas, School of Medicine Kansas City, Kansas George C. -

The Functional Anatomy of the Heart. Development of the Heart, Anomalies
The functional anatomy of the heart. Development of the heart, anomalies Anatomy and Clinical Anatomy Department Anastasia Bendelic Plan: Cardiovascular system – general information Heart – functional anatomy Development of the heart Abnormalities of the heart Examination in a living person Cardiovascular system Cardiovascular system (also known as vascular system, or circulatory system) consists of: 1. heart; 2. blood vessels (arteries, veins, capillaries); 3. lymphatic vessels. Blood vessels Arteries are blood vessels that carry blood away from the heart. Veins carry blood back towards the heart. Capillaries are tiny blood vessels, that connect arteries to veins. Lymphatic vessels: lymphatic capillaries; lymphatic vessels (superficial and deep lymph vessels); lymphatic trunks (jugular, subclavian, bronchomediastinal, lumbar, intestinal trunks); lymphatic ducts (thoracic duct and right lymphatic duct). Lymphatic vessels Microcirculation Microcirculatory bed comprises 7 components: 1. arterioles; 2. precapillaries or precapillary arterioles; 3. capillaries; 4. postcapillaries or postcapillary venules; 5. venules; 6. lymphatic capillaries; 7. interstitial component. Microcirculation The heart Heart is shaped as a pyramid with: an apex (directed downward, forward and to the left); a base (facing upward, backward and to the right). There are four surfaces of the heart: sternocostal (anterior) surface; diaphragmatic (inferior) surface; right pulmonary surface; left pulmonary surface. External surface of the heart The heart The heart has four chambers: right and left atria; right and left ventricles. Externally, the atria are demarcated from the ventricles by coronary sulcus (L. sulcus coronarius). The right and left ventricles are demarcated from each other by anterior and posterior interventricular sulci (L. sulci interventriculares anterior et posterior). Chambers of the heart The atria The atria are thin-walled chambers, that receive blood from the veins and pump it into the ventricles.