Cytogenetic Abnormalities in 772 ... -.: Scientific Press International Limited
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Familial Intellectual Disability As a Result of a Derivative Chromosome
Zhang et al. Molecular Cytogenetics (2018) 11:18 DOI 10.1186/s13039-017-0349-x RESEARCH Open Access Familial intellectual disability as a result of a derivative chromosome 22 originating from a balanced translocation (3;22) in a four generation family Kaihui Zhang1†, Yan Huang2†, Rui Dong1†, Yali Yang2, Ying Wang1, Haiyan Zhang1, Yufeng Zhang1, Zhongtao Gai1* and Yi Liu1* Abstract Background: Balanced reciprocal translocation is usually an exchange of two terminal segments from different chromosomes without phenotypic effect on the carrier while leading to increased risk of generating unbalanced gametes. Here we describe a four-generation family in Shandong province of China with at least three patients sharing severe intellectual disability and developmental delay resulting from a derivative chromosome 22 originating from a balanced translocation (3;22) involving chromosomes 3q28q29 and 22q13.3. Methods: The proband and his relatives were detected by using karyotyping, chromosome microarray analysis, fluorescent in situ hybridization and real-time qPCR. Results: The proband, a 17 month-old boy, presented with severe intellectual disability, developmental delay, specific facial features and special posture of hands. Pedigree analysis showed that there were at least three affected patients. The proband and other two living patients manifested similar phenotypes and were identified to have identically abnormal cytogenetic result with an unbalanced translocation of 9.0 Mb duplication at 3q28q29 and a 1.7Mb microdeletion at 22q13.3 by karyotyping and chromosome microarray analysis. His father and other five relatives had a balanced translocation of 3q and 22q. Fluorescence in situ hybridization and real-time qPCR definitely validated the results. -

3 Chromosome Chapter
Chromosome 3 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_03.gif Introduction The size of chromosome 3 is ~200 Mb. Within this chromosome, there are thousands of genes, many of which are necessary for normal intellectual development or involved in the formation of body organs. Deletions of Chromosome 3 The length of the short arm of chromosome 3 is ~90 Mb. Most known deletions of 3p are caused by the loss of its distal 15 Mb segment (3p25–pter). Deletions of the more proximal segments are relatively rare; there are only ~50 reports on such patients. Therefore, it would be premature to talk about any syndrome related to deletions of the proximal part of 3p. The location of the breakpoints, size of deletion, and reported abnormalities are different in most described patients. However, recurrent aortal stenosis in patients with deletion 3p11p14.2, abnormal lung lobation in patients with deletion 3p12p14.2, agenesis or hypoplasia of the corpus callosum in patients with deletion 3p13, microphthalmia and coloboma in patients with deletion 3p13p21.1, choanal atresia and absent gallbladder in patients with deletion 3p13p21, and hearing loss in patients with deletion 3p14 are all indicators that the above–mentioned segments likely contain genes involved in the formation of these systems. Deletions of 3p Deletion of 3p25–pter The most distal segment of the short arm of chromosome 3 is 3p26 and spans ~8 Mb. -
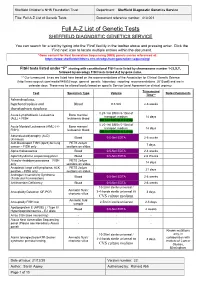
Full A-Z List of Genetic Tests Document Reference Number: 413.001
Sheffield Children’s NHS Foundation Trust Department: Sheffield Diagnostic Genetics Service Title: Full A-Z List of Genetic Tests Document reference number: 413.001 Full A-Z List of Genetic Tests SHEFFIELD DIAGNOSTIC GENETICS SERVICE You can search for a test by typing into the ‘Find’ facility in the toolbar above and pressing enter. Click the ‘Find next’ icon to locate multiple entries within the document. *Gene content for Next Generation Sequencing (NGS) panels can be referenced at: https://www.sheffieldchildrens.nhs.uk/sdgs/next-generation-sequencing/ FISH tests listed under “F” starting with constitutional FISH tests listed by chromosome number 1-22,X,Y, followed by oncology FISH tests listed A-Z by gene name. ** Our turnaround times are listed here based on the recommendations of the Association for Clinical Genetic Science (http://www.acgs.uk.com/media/949852/acgs_general_genetic_laboratory_reporting_recommendations_2015.pdf) and are in calendar days. These may be altered locally based on specific Service Level Agreement or clinical urgency. Turnaround Test Specimen Type Volume Notes/Comments Time** Achondroplasia, hypchondroplasia and Blood 0.5-5ml 2-6 weeks thanatophoric dysplasia 0.25-1ml BM in 5-10ml of Acute Lymphoblastic Leukaemia Bone marrow/ transport medium 14 days (ALL) + FISH leukaemic blood OR 1ml BM/VB in Li Hep 0.25-1ml BM in 5-10ml of Acute Myeloid Leukaemia (AML) (+/- Bone marrow/ transport medium 14 days FISH) leukaemic blood OR 1ml BM/VB in Li Hep Adrenoleukodystrophy (ALD) Blood 0.5-5ml EDTA 2-6 weeks (X-linked) -

See Shalini C Reshmi's Curriculum Vitae
Shalini C. Reshmi, Ph.D. Contact Information: Institute for Genomic Medicine Nationwide Children’s Hospital 575 Childrens Crossroads, RB3-WB2233 Columbus, OH 43215 Lab: (614) 722-5321 Fax: (614) 355-6833 [email protected] CURRENT ACADEMIC POSITIONS 2/2018-present Assistant Professor, Department of Pediatrics, The Ohio State University, Columbus, OH 7/2009-present Assistant Professor (Clinical), Department of Pathology, The Ohio State University, Columbus, OH ADMINISTRATIVE APPOINTMENT 10/2016-present Director, Clinical Laboratory Institute for Genomic Medicine Nationwide Children's Hospital, Columbus, OH 9/2012-10/2016 Associate Director, Cytogenetics/Molecular Genetics Laboratory Nationwide Children's Hospital, Columbus, OH 12/2008-9/2012 Assistant Director, Cytogenetics/Molecular Genetics Laboratory Nationwide Children's Hospital, Columbus, OH EDUCATION 9/2001-5/2005 Ph.D. Human Genetics University of Pittsburgh, Graduate School of Public Health Pittsburgh, PA 9/1994-4/1996 M.S. Human Genetics University of Pittsburgh, Graduate School of Public Health Pittsburgh, PA 8/1988-5/1992 B.S. Biology The Catholic University of America Washington, D.C. TRAINING 7/2007-10/2008 American College of Medical Genetics and Genomics Clinical Molecular Genetics University of Chicago, Pritzker School of Medicine Chicago, IL 7/2005-6/2007 American College of Medical Genetics and Genomics Clinical Cytogenetics University of Chicago, Pritzker School of Medicine Chicago, IL Board Certifications 2 9/01/09- Diplomate, American Board of -

Deletion 2Q24
Deletion 2q24 Authors: Doctor Saskia M. Maas1 Scientific editor: Professor Raoul CM Hennekam Creation date: June 2001 Update: March 2004 1Department of Clinical Genetics, Academic Medical Centre, University of Amsterdam, P.O. Box 22700, 1100 DE Amsterdam. The Netherlands. mailto:S.M. [email protected] Abstract Key words Disease name and synonyms Excluded diseases Diagnosis criteria/definition Differential diagnosis Prevalence Clinical description Management including treatment Etiology Diagnostic methods Genetic counselling Antenatal diagnosis Unresolved questions References Abstract Deletion of 2q24, i.e. the loss of a small but specific region of the long arm of chromosome 2 forms a clinically recognizable syndrome. Twenty-three cases are known from the literature. Several symptoms are commonly seen in patients with chromosome aberrations, such as low birth weight, growth retardation, mental retardation, and low and malformed ears. However, the eye anomalies (coloboma, cataract and microphthalmia), anomalies of hands and feet (flexion deformities) and the congenital heart defects are more specific features, and are especially important in recognizing the clinical phenotype. Key words chromosome 2q23q24, interstitial deletion, camptodactyly, eye anomalies, cardiac defects Disease name and synonyms and feet and/or heart defects seem to be more Deletion of chromosome band 2q24 specific for this deletion. Monosomy 2q24 Differential diagnosis Excluded diseases If the cytogenetic anomaly is detected, no All the other chromosome anomalies associated differential diagnosis is needed. If no with low birth weight, growth retardation, mental microdeletion at 2q24 can be found the clinical retardation and facial dysmorphism. features may point to a vast array of other chromosome anomalies of monogenic entities. Diagnosis criteria/definition The diagnosis is made when chromosome Prevalence analysis shows a deletion of a specific part To our knowledge there are twenty-three (band q24) of the long arm of chromosome 2. -

Chromosome 17
Chromosome 17 Description Humans normally have 46 chromosomes in each cell, divided into 23 pairs. Two copies of chromosome 17, one copy inherited from each parent, form one of the pairs. Chromosome 17 spans about 83 million DNA building blocks (base pairs) and represents between 2.5 and 3 percent of the total DNA in cells. Identifying genes on each chromosome is an active area of genetic research. Because researchers use different approaches to predict the number of genes on each chromosome, the estimated number of genes varies. Chromosome 17 likely contains 1, 100 to 1,200 genes that provide instructions for making proteins. These proteins perform a variety of different roles in the body. Health Conditions Related to Chromosomal Changes The following chromosomal conditions are associated with changes in the structure or number of copies of chromosome 17. 17q12 deletion syndrome 17q12 deletion syndrome is a condition that results from the deletion of a small piece of chromosome 17 in each cell. Signs and symptoms of 17q12 deletion syndrome can include abnormalities of the kidneys and urinary system, a form of diabetes called maturity-onset diabetes of the young type 5 (MODY5), delayed development, intellectual disability, and behavioral or psychiatric disorders. Some females with this chromosomal change have Mayer-Rokitansky-Küster-Hauser syndrome, which is characterized by underdevelopment or absence of the vagina and uterus. Features associated with 17q12 deletion syndrome vary widely, even among affected members of the same family. Most people with 17q12 deletion syndrome are missing about 1.4 million DNA building blocks (base pairs), also written as 1.4 megabases (Mb), on the long (q) arm of the chromosome at a position designated q12. -

Aneuploidy and More in Neural Diversity and Disease
Seminars in Cell & Developmental Biology 24 (2013) 357–369 Contents lists available at SciVerse ScienceDirect Seminars in Cell & Developmental Biology jo urnal homepage: www.elsevier.com/locate/semcdb Review The genomically mosaic brain: Aneuploidy and more in neural diversity and disease a,b a,∗ Diane M. Bushman , Jerold Chun a Molecular and Cellular Neuroscience Department, Dorris Neuroscience Center, The Scripps Research Institute, La Jolla, CA, USA b Biomedical Sciences Graduate Program, School of Medicine, University of California San Diego, La Jolla, CA, USA a r t i c l e i n f o a b s t r a c t Article history: Genomically identical cells have long been assumed to comprise the human brain, with post-genomic Available online 4 March 2013 mechanisms giving rise to its enormous diversity, complexity, and disease susceptibility. However, the identification of neural cells containing somatically generated mosaic aneuploidy – loss and/or gain of Keywords: chromosomes from a euploid complement – and other genomic variations including LINE1 retrotrans- Aneuploidy posons and regional patterns of DNA content variation (DCV), demonstrate that the brain is genomically Genome heterogeneous. The precise phenotypes and functions produced by genomic mosaicism are not well Mosaicism understood, although the effects of constitutive aberrations, as observed in Down syndrome, implicate Somatic mutation CNS roles for defined mosaic genomes relevant to cellular survival, differentiation potential, stem cell biology, CNV and brain organization. Here we discuss genomic mosaicism as a feature of the normal brain as well as a possible factor in the weak or complex genetic linkages observed for many of the most common forms of neurological and psychiatric diseases. -

In Two Patients with 16P11.2 Microdeletion Syndrome
European Journal of Human Genetics (2012), 1–5 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Childhood Apraxia of Speech (CAS) in two patients with 16p11.2 microdeletion syndrome Gordana Raca1,2, Becky S Baas3, Salman Kirmani4, Jennifer J Laffin1,2, Craig A Jackson2, Edythe A Strand3, Kathy J Jakielski5 and Lawrence D Shriberg1,6 We report clinical findings that extend the phenotype of the B550 kb 16p11.2 microdeletion syndrome to include a rare, severe, and persistent pediatric speech sound disorder termed Childhood Apraxia of Speech (CAS). CAS is the speech disorder identified in a multigenerational pedigree (‘KE’) in which half of the members have a mutation in FOXP2 that co-segregates with CAS, oromotor apraxia, and low scores on a nonword repetition task. Each of the two patients in the current report completed a 2-h assessment protocol that provided information on their cognitive, language, speech, oral mechanism, motor, and developmental histories and performance. Their histories and standard scores on perceptual and acoustic speech tasks met clinical and research criteria for CAS. Array comparative genomic hybridization analyses identified deletions at chromosome 16p11.2 in each patient. These are the first reported cases with well-characterized CAS in the 16p11.2 syndrome literature and the first report of this microdeletion in CAS genetics research. We discuss implications of findings for issues in both literatures. European Journal of Human Genetics advance online publication, 22 -

Verifi Plus Handout
Verifi™ Plus Prenatal Test Screen for a broader range of chromosomal aneuploidies The new all-chromosome option of the Verifi Plus Prenatal Test will provide information about trisomies for all chromosomes, giving you and your patients a broader range of information than the standard Verifi Prenatal Test. This option within the Verifi Plus Prenatal Test, allows screening for rare autosomal trisomies that may occur, especially in the presence of an abnormal ultrasound. Guidelines recommend invasive diagnostic follow-up for those patients with ultrasound anomalies; however, for those who decline invasive diagnostic follow-up, the all-chromosome screening test could be an option. Chromosomal aneuploidies, in general, may lead to varying degrees of structural defects, and developmental and intellectual disabilities.1 Chromosomal aneuploidies identified with this test may be representative of the chromosomal make up of every fetal cell (full fetal aneuploidy), some fetal cells (fetal mosaicism), placental cells only (confined placental mosaicism), or some maternal cells (maternal mosaicism). The clinical significance of rare chromosome aneuploidies is variable and depends on the specific finding and which cells are involved. Patients with a positive NIPT result should be offered further detailed counseling and diagnostic testing such as chorionic villus sampling or amniocentesis.2 Extensive options for more personalized screening The Verifi Plus Prenatal Test offers the following testing options: • Aneuploidy of chromosomes 21, 18, and 13 (trisomy 21, 18, and 13) is always included. • Sex chromosome aneuploidies (monosomy X, XXX, XXY, and XYY) are included if requested; fetal sex (XX or XY) will be reported if no sex chromosome aneuploidy is detected. -

Medical Sciences
medical sciences Article Integrated FISH, Karyotyping and aCGH Analyses for Effective Prenatal Diagnosis of Common Aneuploidies and Other Cytogenomic Abnormalities Hongyan Chai, Autumn DiAdamo, Brittany Grommisch, Jennifer Boyle, Katherine Amato, Dongmei Wang, Jiadi Wen and Peining Li * Laboratory of Clinical Cytogenetics, Department of Genetics, Yale School of Medicine, New Haven, CT 06520, USA; [email protected] (H.C.); [email protected] (A.D.); [email protected] (B.G.); [email protected] (J.B.); [email protected] (K.A.); [email protected] (D.W.); [email protected] (J.W.) * Correspondence: [email protected]; Tel.: +1-203-785-6317; Fax: +1-203-785-7342 Received: 13 December 2018; Accepted: 21 January 2019; Published: 23 January 2019 Abstract: Current prenatal genetic evaluation showed a significantly increase in non-invasive screening and the reduction of invasive diagnostic procedures. To evaluate the diagnostic efficacy on detecting common aneuploidies, structural chromosomal rearrangements, and pathogenic copy number variants (pCNV), we performed a retrospective analysis on a case series initially analyzed by aneuvysion fluorescence in situ hybridization (FISH) and karyotyping then followed by array comparative genomic hybridization (aCGH). Of the 386 cases retrieved from the past decade, common aneuploidies were detected in 137 cases (35.5%), other chromosomal structural rearrangements were detected in four cases (1%), and pCNV were detected in five cases (1.3%). The relative frequencies for common aneuploidies suggested an under detection of sex chromosome aneuploidies. Approximately 9.5% of cases with common aneuploidies showed a mosaic pattern. Inconsistent results between FISH and karyotyping were noted in cases with pseudo-mosaicism introduced by culture artifact or variable cellular proliferation from cells with mosaic karyotypic complements under in vitro cell culture. -

Dissecting Molecular Genetic Mechanisms of 1Q21.1 CNV in Neuropsychiatric Disorders
International Journal of Molecular Sciences Review Dissecting Molecular Genetic Mechanisms of 1q21.1 CNV in Neuropsychiatric Disorders Joy Yoon and Yingwei Mao * Department of Biology, Eberly College of Science, Pennsylvania State University, University Park, PA 16802, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-814-867-4739 Abstract: Pathogenic copy number variations (CNVs) contribute to the etiology of neurodevelopmen- tal/neuropsychiatric disorders (NDs). Increased CNV burden has been found to be critically involved in NDs compared with controls in clinical studies. The 1q21.1 CNVs, rare and large chromosomal microduplications and microdeletions, are detected in many patients with NDs. Phenotypes of duplication and deletion appear at the two ends of the spectrum. Microdeletions are predominant in individuals with schizophrenia (SCZ) and microcephaly, whereas microduplications are predominant in individuals with autism spectrum disorder (ASD) and macrocephaly. However, its complexity hinders the discovery of molecular pathways and phenotypic networks. In this review, we summarize the recent genome-wide association studies (GWASs) that have identified candidate genes positively correlated with 1q21.1 CNVs, which are likely to contribute to abnormal phenotypes in carriers. We discuss the clinical data implicated in the 1q21.1 genetic structure that is strongly associated with neurodevelopmental dysfunctions like cognitive impairment and reduced synaptic plasticity. We further present variations reported in the phenotypic severity, genomic penetrance and inheritance. Keywords: copy number variation; microdeletion; microduplication; schizophrenia; autism spectrum Citation: Yoon, J.; Mao, Y. Dissecting disorder; microcephaly; macrocephaly; neurodegeneration; synaptic plasticity Molecular Genetic Mechanisms of 1q21.1 CNV in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2021, 22, 5811. https://doi.org/10.3390/ 1. -

Chromosome 5
Chromosome 5 Description Humans normally have 46 chromosomes in each cell, divided into 23 pairs. Two copies of chromosome 5, one copy inherited from each parent, form one of the pairs. Chromosome 5 spans about 181 million DNA building blocks (base pairs) and represents almost 6 percent of the total DNA in cells. Identifying genes on each chromosome is an active area of genetic research. Because researchers use different approaches to predict the number of genes on each chromosome, the estimated number of genes varies. Chromosome 5 likely contains about 900 genes that provide instructions for making proteins. These proteins perform a variety of different roles in the body. Health Conditions Related to Chromosomal Changes The following chromosomal conditions are associated with changes in the structure or number of copies of chromosome 5. 5q minus syndrome Deletion of a region of DNA from the long (q) arm of chromosome 5 is involved in a condition called 5q minus (5q-) syndrome. This deletion occurs in immature blood cells during a person's lifetime and affects one copy of chromosome 5 in each cell. 5q- syndrome is a type of bone marrow disorder called myelodysplastic syndrome (MDS), in which immature blood cells fail to develop normally. Individuals with 5q- syndrome often have a shortage of red blood cells (anemia) and abnormalities in blood cells called megakaryocytes, which produce platelets, the cells involved in blood clotting. Affected individuals also have an increased risk of developing a fast-growing blood cancer known as acute myeloid leukemia (AML). Most people with 5q- syndrome are missing a sequence of about 1.5 million DNA base pairs, also written as 1.5 megabases (Mb).