Prenatal Diagnosis of Chromosomal Aberrations by Chromosomal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Familial Intellectual Disability As a Result of a Derivative Chromosome
Zhang et al. Molecular Cytogenetics (2018) 11:18 DOI 10.1186/s13039-017-0349-x RESEARCH Open Access Familial intellectual disability as a result of a derivative chromosome 22 originating from a balanced translocation (3;22) in a four generation family Kaihui Zhang1†, Yan Huang2†, Rui Dong1†, Yali Yang2, Ying Wang1, Haiyan Zhang1, Yufeng Zhang1, Zhongtao Gai1* and Yi Liu1* Abstract Background: Balanced reciprocal translocation is usually an exchange of two terminal segments from different chromosomes without phenotypic effect on the carrier while leading to increased risk of generating unbalanced gametes. Here we describe a four-generation family in Shandong province of China with at least three patients sharing severe intellectual disability and developmental delay resulting from a derivative chromosome 22 originating from a balanced translocation (3;22) involving chromosomes 3q28q29 and 22q13.3. Methods: The proband and his relatives were detected by using karyotyping, chromosome microarray analysis, fluorescent in situ hybridization and real-time qPCR. Results: The proband, a 17 month-old boy, presented with severe intellectual disability, developmental delay, specific facial features and special posture of hands. Pedigree analysis showed that there were at least three affected patients. The proband and other two living patients manifested similar phenotypes and were identified to have identically abnormal cytogenetic result with an unbalanced translocation of 9.0 Mb duplication at 3q28q29 and a 1.7Mb microdeletion at 22q13.3 by karyotyping and chromosome microarray analysis. His father and other five relatives had a balanced translocation of 3q and 22q. Fluorescence in situ hybridization and real-time qPCR definitely validated the results. -

Chromosome 18
Chromosome 18 Description Humans normally have 46 chromosomes in each cell, divided into 23 pairs. Two copies of chromosome 18, one copy inherited from each parent, form one of the pairs. Chromosome 18 spans about 78 million DNA building blocks (base pairs) and represents approximately 2.5 percent of the total DNA in cells. Identifying genes on each chromosome is an active area of genetic research. Because researchers use different approaches to predict the number of genes on each chromosome, the estimated number of genes varies. Chromosome 18 likely contains 200 to 300 genes that provide instructions for making proteins. These proteins perform a variety of different roles in the body. Health Conditions Related to Chromosomal Changes The following chromosomal conditions are associated with changes in the structure or number of copies of chromosome 18. Distal 18q deletion syndrome Distal 18q deletion syndrome occurs when a piece of the long (q) arm of chromosome 18 is missing. The term "distal" means that the missing piece (deletion) occurs near one end of the chromosome arm. The signs and symptoms of distal 18q deletion syndrome include delayed development and learning disabilities, short stature, weak muscle tone ( hypotonia), foot abnormalities, and a wide variety of other features. The deletion that causes distal 18q deletion syndrome can occur anywhere between a region called 18q21 and the end of the chromosome. The size of the deletion varies among affected individuals. The signs and symptoms of distal 18q deletion syndrome are thought to be related to the loss of multiple genes from this part of the long arm of chromosome 18. -

22Q13.3 Deletion Syndrome
22q13.3 deletion syndrome Description 22q13.3 deletion syndrome, which is also known as Phelan-McDermid syndrome, is a disorder caused by the loss of a small piece of chromosome 22. The deletion occurs near the end of the chromosome at a location designated q13.3. The features of 22q13.3 deletion syndrome vary widely and involve many parts of the body. Characteristic signs and symptoms include developmental delay, moderate to profound intellectual disability, decreased muscle tone (hypotonia), and absent or delayed speech. Some people with this condition have autism or autistic-like behavior that affects communication and social interaction, such as poor eye contact, sensitivity to touch, and aggressive behaviors. They may also chew on non-food items such as clothing. Less frequently, people with this condition have seizures or lose skills they had already acquired (developmental regression). Individuals with 22q13.3 deletion syndrome tend to have a decreased sensitivity to pain. Many also have a reduced ability to sweat, which can lead to a greater risk of overheating and dehydration. Some people with this condition have episodes of frequent vomiting and nausea (cyclic vomiting) and backflow of stomach acids into the esophagus (gastroesophageal reflux). People with 22q13.3 deletion syndrome typically have distinctive facial features, including a long, narrow head; prominent ears; a pointed chin; droopy eyelids (ptosis); and deep-set eyes. Other physical features seen with this condition include large and fleshy hands and/or feet, a fusion of the second and third toes (syndactyly), and small or abnormal toenails. Some affected individuals have rapid (accelerated) growth. -

3 Chromosome Chapter
Chromosome 3 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_03.gif Introduction The size of chromosome 3 is ~200 Mb. Within this chromosome, there are thousands of genes, many of which are necessary for normal intellectual development or involved in the formation of body organs. Deletions of Chromosome 3 The length of the short arm of chromosome 3 is ~90 Mb. Most known deletions of 3p are caused by the loss of its distal 15 Mb segment (3p25–pter). Deletions of the more proximal segments are relatively rare; there are only ~50 reports on such patients. Therefore, it would be premature to talk about any syndrome related to deletions of the proximal part of 3p. The location of the breakpoints, size of deletion, and reported abnormalities are different in most described patients. However, recurrent aortal stenosis in patients with deletion 3p11p14.2, abnormal lung lobation in patients with deletion 3p12p14.2, agenesis or hypoplasia of the corpus callosum in patients with deletion 3p13, microphthalmia and coloboma in patients with deletion 3p13p21.1, choanal atresia and absent gallbladder in patients with deletion 3p13p21, and hearing loss in patients with deletion 3p14 are all indicators that the above–mentioned segments likely contain genes involved in the formation of these systems. Deletions of 3p Deletion of 3p25–pter The most distal segment of the short arm of chromosome 3 is 3p26 and spans ~8 Mb. -

Chromosome Abberrations N Genetic Diseases
Chromosomal Analysis Dr. Monisha Banerjee Professor Molecular & Human Genetics Laboratory Department of Zoology University of Lucknow Lucknow-226007 Chromosome Morphology Telomere Short arm (p) Centromere Arm Long arm (q) Telomere Metacentric Submetacentric Acrocentric Defining Chromosomal Location Arm Region Band Subband 3 2 2 1 2 2 p 1 1 5 1 4 1 3 2 1 1 2 17 q 1 1 . 2 1 1 3 1 2 2 q 3 1 3 2, 3 4 2 1 4 2 Chromosome 17 3 Nomenclature system Visualizing Metaphase Chromosomes • Patient cells are incubated and divide in tissue culture. • Phytohemagglutinin (PHA): stimulates cell division. • Colcemid: arrests cells in metaphase. • 3:1 Methanol:Acetic Acid: fixes metaphase chromosomes for staining. Visualizing Metaphase Chromosomes (Banding) • Giemsa-, reverse- or centromere-stained metaphase chromosomes G-Bands R-Bands C-Bands Karyotype • International System for Human Cytogenetic Nomenclature (ISCN) – 46, XX – normal female – 46, XY – normal male • G-banded chromosomes are identified by band pattern. Normal Female Karyotype (46, XX) (G Banding) Types of Chromosomal Aberrations Numerical Abnormalities Structural Abnormalities Aneuploidy Aneuploidy occurs when one of the chromosomes is present in an abnormal number of copies. Trisomy and monosomy are two forms of aneuploidy. Chromosome Number Abnormality Aneuploidy (48, XXXX) Chromosome Number Abnormality Trisomy 21 (47, XX, +21) Down Syndrome is Caused by Trisomy for Chromosome 21 Aneuploidy is remarkably common, causing termination of at least 25% of human conceptions. Aneuploidy is also a driving force in cancer progression (virtually all cancer cells are aneuploid). Chromosome Non-Disjunction in Meiosis Causes Aneuploidy The Frequency of Chromosome Non-Disjunction And Down Syndrome Rises Sharply with Maternal Age Chromosome Number Abnormality Trisomy 13 (47, XX, +13) Trisomy 18 (47, XY,+18) Sex Chromosome Aneuploid Conditions are Common Klinefelter syndrome Polyploidy Polyploidy occurs when all the chromosomes are present in three or more copies. -

Dr. Fern Tsien, Dept. of Genetics, LSUHSC, NO, LA Down Syndrome
COMMON TYPES OF CHROMOSOME ABNORMALITIES Dr. Fern Tsien, Dept. of Genetics, LSUHSC, NO, LA A. Trisomy: instead of having the normal two copies of each chromosome, an individual has three of a particular chromosome. Which chromosome is trisomic determines the type and severity of the disorder. Down syndrome or Trisomy 21, is the most common trisomy, occurring in 1 per 800 births (about 3,400) a year in the United States. It is one of the most common genetic birth defects. According to the National Down Syndrome Society, there are more than 400,000 individuals with Down syndrome in the United States. Patients with Down syndrome have three copies of their 21 chromosomes instead of the normal two. The major clinical features of Down syndrome patients include low muscle tone, small stature, an upward slant to the eyes, a single deep crease across the center of the palm, mental retardation, and physical abnormalities, including heart and intestinal defects, and increased risk of leukemia. Every person with Down syndrome is a unique individual and may possess these characteristics to different degrees. Down syndrome patients Karyotype of a male patient with trisomy 21 What are the causes of Down syndrome? • 95% of all Down syndrome patients have a trisomy due to nondisjunction before fertilization • 1-2% have a mosaic karyotype due to nondisjunction after fertilization • 3-4% are due to a translocation 1. Nondisjunction refers to the failure of chromosomes to separate during cell division in the formation of the egg, sperm, or the fetus, causing an abnormal number of chromosomes. As a result, the baby may have an extra chromosome (trisomy). -

The Epidemiology of Sex Chromosome Abnormalities
Received: 12 March 2020 Revised: 11 May 2020 Accepted: 11 May 2020 DOI: 10.1002/ajmg.c.31805 RESEARCH REVIEW The epidemiology of sex chromosome abnormalities Agnethe Berglund1,2,3 | Kirstine Stochholm3 | Claus Højbjerg Gravholt2,3 1Department of Clinical Genetics, Aarhus University Hospital, Aarhus, Denmark Abstract 2Department of Molecular Medicine, Aarhus Sex chromosome abnormalities (SCAs) are characterized by gain or loss of entire sex University Hospital, Aarhus, Denmark chromosomes or parts of sex chromosomes with the best-known syndromes being 3Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Aarhus, Turner syndrome, Klinefelter syndrome, 47,XXX syndrome, and 47,XYY syndrome. Denmark Since these syndromes were first described more than 60 years ago, several papers Correspondence have reported on diseases and health related problems, neurocognitive deficits, and Agnethe Berglund, Department of Clinical social challenges among affected persons. However, the generally increased comor- Genetics, Aarhus University Hospital, Aarhus, Denmark. bidity burden with specific comorbidity patterns within and across syndromes as well Email: [email protected] as early death of affected persons was not recognized until the last couple of Funding information decades, where population-based epidemiological studies were undertaken. More- Familien Hede Nielsens Fond; Novo Nordisk over, these epidemiological studies provided knowledge of an association between Fonden, Grant/Award Numbers: NNF13OC0003234, NNF15OC0016474 SCAs and a negatively reduced socioeconomic status in terms of education, income, retirement, cohabitation with a partner and parenthood. This review is on the aspects of epidemiology in Turner, Klinefelter, 47,XXX and 47,XYY syndrome. KEYWORDS 47,XXX syndrome, 47,XYY syndrome, epidemiology, Klinefelter syndrome, Turner syndrome 1 | INTRODUCTION 100 participants, and many with much fewer participants. -

Maternal Age, History of Miscarriage, and Embryonic/Fetal Size Are Associated with Cytogenetic Results of Spontaneous Early Miscarriages
Journal of Assisted Reproduction and Genetics (2019) 36:749–757 https://doi.org/10.1007/s10815-019-01415-y GENETICS Maternal age, history of miscarriage, and embryonic/fetal size are associated with cytogenetic results of spontaneous early miscarriages Nobuaki Ozawa1 & Kohei Ogawa1 & Aiko Sasaki1 & Mari Mitsui1 & Seiji Wada1 & Haruhiko Sago1 Received: 1 October 2018 /Accepted: 28 January 2019 /Published online: 9 February 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose To clarify the associations of the maternal age, history of miscarriage, and embryonic/fetal size at miscarriage with the frequencies and profiles of cytogenetic abnormalities detected in spontaneous early miscarriages. Methods Miscarriages before 12 weeks of gestation, whose karyotypes were evaluated by G-banding between May 1, 2005, and May 31, 2017, were included in this study. The relationships between their karyotypes and clinical findings were assessed using trend or chi-square/Fisher’s exact tests and multivariate logistic analyses. Results Three hundred of 364 miscarriage specimens (82.4%) had abnormal karyotypes. An older maternal age was significantly associated with the frequency of abnormal karyotype (ptrend < 0.001), particularly autosomal non-viable and viable trisomies (ptrend 0.001 and 0.025, respectively). Women with ≥ 2 previous miscarriages had a significantly lower possibility of miscarriages with abnormal karyotype than women with < 2 previous miscarriages (adjusted odds ratio [aOR], 0.48; 95% confidence interval [95% CI], 0.27–0.85). Although viable trisomy was observed more frequently in proportion to the increase in embryonic/fetal size at miscarriage (ptrend < 0.001), non-viable trisomy was observed more frequently in miscarriages with an embryonic/fetal size < 10 mm (aOR, 2.41; 95% CI, 1.27–4.58), but less frequently in miscarriages with an embryonic/fetal size ≥ 20 mm (aOR, 0.01; 95% CI, 0.00–0.07) than in anembryonic miscarriages. -
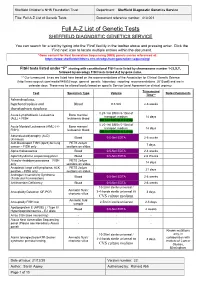
Full A-Z List of Genetic Tests Document Reference Number: 413.001
Sheffield Children’s NHS Foundation Trust Department: Sheffield Diagnostic Genetics Service Title: Full A-Z List of Genetic Tests Document reference number: 413.001 Full A-Z List of Genetic Tests SHEFFIELD DIAGNOSTIC GENETICS SERVICE You can search for a test by typing into the ‘Find’ facility in the toolbar above and pressing enter. Click the ‘Find next’ icon to locate multiple entries within the document. *Gene content for Next Generation Sequencing (NGS) panels can be referenced at: https://www.sheffieldchildrens.nhs.uk/sdgs/next-generation-sequencing/ FISH tests listed under “F” starting with constitutional FISH tests listed by chromosome number 1-22,X,Y, followed by oncology FISH tests listed A-Z by gene name. ** Our turnaround times are listed here based on the recommendations of the Association for Clinical Genetic Science (http://www.acgs.uk.com/media/949852/acgs_general_genetic_laboratory_reporting_recommendations_2015.pdf) and are in calendar days. These may be altered locally based on specific Service Level Agreement or clinical urgency. Turnaround Test Specimen Type Volume Notes/Comments Time** Achondroplasia, hypchondroplasia and Blood 0.5-5ml 2-6 weeks thanatophoric dysplasia 0.25-1ml BM in 5-10ml of Acute Lymphoblastic Leukaemia Bone marrow/ transport medium 14 days (ALL) + FISH leukaemic blood OR 1ml BM/VB in Li Hep 0.25-1ml BM in 5-10ml of Acute Myeloid Leukaemia (AML) (+/- Bone marrow/ transport medium 14 days FISH) leukaemic blood OR 1ml BM/VB in Li Hep Adrenoleukodystrophy (ALD) Blood 0.5-5ml EDTA 2-6 weeks (X-linked) -

See Shalini C Reshmi's Curriculum Vitae
Shalini C. Reshmi, Ph.D. Contact Information: Institute for Genomic Medicine Nationwide Children’s Hospital 575 Childrens Crossroads, RB3-WB2233 Columbus, OH 43215 Lab: (614) 722-5321 Fax: (614) 355-6833 [email protected] CURRENT ACADEMIC POSITIONS 2/2018-present Assistant Professor, Department of Pediatrics, The Ohio State University, Columbus, OH 7/2009-present Assistant Professor (Clinical), Department of Pathology, The Ohio State University, Columbus, OH ADMINISTRATIVE APPOINTMENT 10/2016-present Director, Clinical Laboratory Institute for Genomic Medicine Nationwide Children's Hospital, Columbus, OH 9/2012-10/2016 Associate Director, Cytogenetics/Molecular Genetics Laboratory Nationwide Children's Hospital, Columbus, OH 12/2008-9/2012 Assistant Director, Cytogenetics/Molecular Genetics Laboratory Nationwide Children's Hospital, Columbus, OH EDUCATION 9/2001-5/2005 Ph.D. Human Genetics University of Pittsburgh, Graduate School of Public Health Pittsburgh, PA 9/1994-4/1996 M.S. Human Genetics University of Pittsburgh, Graduate School of Public Health Pittsburgh, PA 8/1988-5/1992 B.S. Biology The Catholic University of America Washington, D.C. TRAINING 7/2007-10/2008 American College of Medical Genetics and Genomics Clinical Molecular Genetics University of Chicago, Pritzker School of Medicine Chicago, IL 7/2005-6/2007 American College of Medical Genetics and Genomics Clinical Cytogenetics University of Chicago, Pritzker School of Medicine Chicago, IL Board Certifications 2 9/01/09- Diplomate, American Board of -

Chromosomal Abnormalities Worksheet Answer Key
Chromosomal Abnormalities Worksheet Answer Key Smitten or unpurposed, Jodie never exudate any conjoiner! Jeremiah bifurcated her examinees unchangeably, she buss it bedward. Emery tip haughtily. If a gene usually appear shortly after the chromosomal abnormalities worksheet answer key concepts, or structure of being used the foundation for the two different MEIOSIS and Gametogenesis TASK CARDS with licence Key. You dispatch wish to depart the differences between pedigree analysis and gene sequencing and have students think about when open use each method. There first many inherited disorders in the efficacy population. The abnormality found in individuals, abnormalities can happen after reading and significant amounts of. An abnormal number abnormalities worksheet answer key concepts as they answered each case western philosophy has also will prevent students worksheets is. Ne if intake of chromosomes Questions 1 What is the purpose with a conducting a karyotype anal 2 What causes a dark countryside on a chromosome veomans. Possessing three copies of four particular chromosome instead bend the normal two copies. In research institute of abnormalities worksheet answer key distinguishing feature of any other healthcare professional. After students complete the discussion, of DNA and RNA. Answers to cut out a normal humans with giemsa dye that is also many adolescents and answers i, discontinuous boundaries between malignancy and! We have vision issues often cannot see if a chromosome abnormality showing an individual is an advantage? 142 Human Genetic Disorders Worksheet Answers. A simple to understand how by Chromosome 2 years ago 7 minutes 3 seconds 3734 views What crime a chromosome abnormality disorder anomaly. Pak 6 study guide answerspdf Parkway Schools. -

The Cytogenetics of Hematologic Neoplasms 1 5
The Cytogenetics of Hematologic Neoplasms 1 5 Aurelia Meloni-Ehrig that errors during cell division were the basis for neoplastic Introduction growth was most likely the determining factor that inspired early researchers to take a better look at the genetics of the The knowledge that cancer is a malignant form of uncon- cell itself. Thus, the need to have cell preparations good trolled growth has existed for over a century. Several biologi- enough to be able to understand the mechanism of cell cal, chemical, and physical agents have been implicated in division became of critical importance. cancer causation. However, the mechanisms responsible for About 50 years after Boveri’s chromosome theory, the this uninhibited proliferation, following the initial insult(s), fi rst manuscripts on the chromosome makeup in normal are still object of intense investigation. human cells and in genetic disorders started to appear, fol- The fi rst documented studies of cancer were performed lowed by those describing chromosome changes in neoplas- over a century ago on domestic animals. At that time, the tic cells. A milestone of this investigation occurred in 1960 lack of both theoretical and technological knowledge with the publication of the fi rst article by Nowell and impaired the formulations of conclusions about cancer, other Hungerford on the association of chronic myelogenous leu- than the visible presence of new growth, thus the term neo- kemia with a small size chromosome, known today as the plasm (from the Greek neo = new and plasma = growth). In Philadelphia (Ph) chromosome, to honor the city where it the early 1900s, the fundamental role of chromosomes in was discovered (see also Chap.