Medical Sciences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accurate Annotation of Human Protein-Coding Small Open Reading Frames
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Nat Chem Manuscript Author Biol. Author Manuscript Author manuscript; available in PMC 2020 June 09. Published in final edited form as: Nat Chem Biol. 2020 April ; 16(4): 458–468. doi:10.1038/s41589-019-0425-0. Accurate annotation of human protein-coding small open reading frames Thomas F. Martinez1,*, Qian Chu1, Cynthia Donaldson1, Dan Tan1, Maxim N. Shokhirev2, Alan Saghatelian1,* 1Clayton Foundation Laboratories for Peptide Biology, Salk Institute for Biological Studies, La Jolla, California 92037, USA 2Razavi Newman Integrative Genomics Bioinformatics Core, Salk Institute for Biological Studies, La Jolla, California 92037, USA Abstract Functional protein-coding small open reading frames (smORFs) are emerging as an important class of genes. However, the number of translated smORFs in the human genome is unclear because proteogenomic methods are not sensitive enough, and, as we show, Ribo-Seq strategies require additional measures to ensure comprehensive and accurate smORF annotation. Here, we integrate de novo transcriptome assembly and Ribo-Seq into an improved workflow that overcomes obstacles with previous methods to more confidently annotate thousands of smORFs. Evolutionary conservation analyses suggest that hundreds of smORF-encoded microproteins are likely functional. Additionally, many smORFs are regulated during fundamental biological processes, such as cell stress. Peptides derived from smORFs are also detectable on human leukocyte antigen complexes, revealing smORFs as a source of antigens. Thus, by including additional validation into our smORF annotation workflow, we accurately identify thousands of unannotated translated smORFs that will provide a rich pool of unexplored, functional human genes. Annotation of open reading frames (ORFs) from genome sequencing was initially carried out by locating in-frame start (AUG) and stop codons1–3. -

Familial Intellectual Disability As a Result of a Derivative Chromosome
Zhang et al. Molecular Cytogenetics (2018) 11:18 DOI 10.1186/s13039-017-0349-x RESEARCH Open Access Familial intellectual disability as a result of a derivative chromosome 22 originating from a balanced translocation (3;22) in a four generation family Kaihui Zhang1†, Yan Huang2†, Rui Dong1†, Yali Yang2, Ying Wang1, Haiyan Zhang1, Yufeng Zhang1, Zhongtao Gai1* and Yi Liu1* Abstract Background: Balanced reciprocal translocation is usually an exchange of two terminal segments from different chromosomes without phenotypic effect on the carrier while leading to increased risk of generating unbalanced gametes. Here we describe a four-generation family in Shandong province of China with at least three patients sharing severe intellectual disability and developmental delay resulting from a derivative chromosome 22 originating from a balanced translocation (3;22) involving chromosomes 3q28q29 and 22q13.3. Methods: The proband and his relatives were detected by using karyotyping, chromosome microarray analysis, fluorescent in situ hybridization and real-time qPCR. Results: The proband, a 17 month-old boy, presented with severe intellectual disability, developmental delay, specific facial features and special posture of hands. Pedigree analysis showed that there were at least three affected patients. The proband and other two living patients manifested similar phenotypes and were identified to have identically abnormal cytogenetic result with an unbalanced translocation of 9.0 Mb duplication at 3q28q29 and a 1.7Mb microdeletion at 22q13.3 by karyotyping and chromosome microarray analysis. His father and other five relatives had a balanced translocation of 3q and 22q. Fluorescence in situ hybridization and real-time qPCR definitely validated the results. -

3 Chromosome Chapter
Chromosome 3 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_03.gif Introduction The size of chromosome 3 is ~200 Mb. Within this chromosome, there are thousands of genes, many of which are necessary for normal intellectual development or involved in the formation of body organs. Deletions of Chromosome 3 The length of the short arm of chromosome 3 is ~90 Mb. Most known deletions of 3p are caused by the loss of its distal 15 Mb segment (3p25–pter). Deletions of the more proximal segments are relatively rare; there are only ~50 reports on such patients. Therefore, it would be premature to talk about any syndrome related to deletions of the proximal part of 3p. The location of the breakpoints, size of deletion, and reported abnormalities are different in most described patients. However, recurrent aortal stenosis in patients with deletion 3p11p14.2, abnormal lung lobation in patients with deletion 3p12p14.2, agenesis or hypoplasia of the corpus callosum in patients with deletion 3p13, microphthalmia and coloboma in patients with deletion 3p13p21.1, choanal atresia and absent gallbladder in patients with deletion 3p13p21, and hearing loss in patients with deletion 3p14 are all indicators that the above–mentioned segments likely contain genes involved in the formation of these systems. Deletions of 3p Deletion of 3p25–pter The most distal segment of the short arm of chromosome 3 is 3p26 and spans ~8 Mb. -

Sema4 Noninvasive Prenatal Select
Sema4 Noninvasive Prenatal Select Noninvasive prenatal testing with targeted genome counting 2 Autosomal trisomies 5 Trisomy 21 (Down syndrome) 6 Trisomy 18 (Edwards syndrome) 7 Trisomy 13 (Patau syndrome) 8 Trisomy 16 9 Trisomy 22 9 Trisomy 15 10 Sex chromosome aneuploidies 12 Monosomy X (Turner syndrome) 13 XXX (Trisomy X) 14 XXY (Klinefelter syndrome) 14 XYY 15 Microdeletions 17 22q11.2 deletion 18 1p36 deletion 20 4p16 deletion (Wolf-Hirschhorn syndrome) 20 5p15 deletion (Cri-du-chat syndrome) 22 15q11.2-q13 deletion (Angelman syndrome) 22 15q11.2-q13 deletion (Prader-Willi syndrome) 24 11q23 deletion (Jacobsen Syndrome) 25 8q24 deletion (Langer-Giedion syndrome) 26 Turnaround time 27 Specimen and shipping requirements 27 2 Noninvasive prenatal testing with targeted genome counting Sema4’s Noninvasive Prenatal Testing (NIPT)- Targeted Genome Counting analyzes genetic information of cell-free DNA (cfDNA) through a simple maternal blood draw to determine the risk for common aneuploidies, sex chromosomal abnormalities, and microdeletions, in addition to fetal gender, as early as nine weeks gestation. The test uses paired-end next-generation sequencing technology to provide higher depth across targeted regions. It also uses a laboratory-specific statistical model to help reduce false positive and false negative rates. The test can be offered to all women with singleton, twins and triplet pregnancies, including egg donor. The conditions offered are shown in below tables. For multiple gestation pregnancies, screening of three conditions -

Predicting Gene Ontology Biological Process from Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R
Methods Predicting Gene Ontology Biological Process From Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R. Hvidsten,2 Herman Midelfart,2 Jan Komorowski,2,3,4 and Arne K. Sandvik1 1Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, N-7489 Trondheim, Norway; 2Department of Information and Computer Science, Norwegian University of Science and Technology, N-7491 Trondheim, Norway; 3The Linnaeus Centre for Bioinformatics, Uppsala University, SE-751 24 Uppsala, Sweden The aim of the present study was to generate hypotheses on the involvement of uncharacterized genes in biological processes. To this end,supervised learning was used to analyz e microarray-derived time-series gene expression data. Our method was objectively evaluated on known genes using cross-validation and provided high-precision Gene Ontology biological process classifications for 211 of the 213 uncharacterized genes in the data set used. In addition,new roles in biological process were hypothesi zed for known genes. Our method uses biological knowledge expressed by Gene Ontology and generates a rule model associating this knowledge with minimal characteristic features of temporal gene expression profiles. This model allows learning and classification of multiple biological process roles for each gene and can predict participation of genes in a biological process even though the genes of this class exhibit a wide variety of gene expression profiles including inverse coregulation. A considerable number of the hypothesized new roles for known genes were confirmed by literature search. In addition,many biological process roles hypothesi zed for uncharacterized genes were found to agree with assumptions based on homology information. -

RNA Editing at Baseline and Following Endoplasmic Reticulum Stress
RNA Editing at Baseline and Following Endoplasmic Reticulum Stress By Allison Leigh Richards A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Human Genetics) in The University of Michigan 2015 Doctoral Committee: Professor Vivian G. Cheung, Chair Assistant Professor Santhi K. Ganesh Professor David Ginsburg Professor Daniel J. Klionsky Dedication To my father, mother, and Matt without whom I would never have made it ii Acknowledgements Thank you first and foremost to my dissertation mentor, Dr. Vivian Cheung. I have learned so much from you over the past several years including presentation skills such as never sighing and never saying “as you can see…” You have taught me how to think outside the box and how to create and explain my story to others. I would not be where I am today without your help and guidance. Thank you to the members of my dissertation committee (Drs. Santhi Ganesh, David Ginsburg and Daniel Klionsky) for all of your advice and support. I would also like to thank the entire Human Genetics Program, and especially JoAnn Sekiguchi and Karen Grahl, for welcoming me to the University of Michigan and making my transition so much easier. Thank you to Michael Boehnke and the Genome Science Training Program for supporting my work. A very special thank you to all of the members of the Cheung lab, past and present. Thank you to Xiaorong Wang for all of your help from the bench to advice on my career. Thank you to Zhengwei Zhu who has helped me immensely throughout my thesis even through my panic. -

Mammalian Protein Expression and Characterization Tools for Next Generation Biologics
Mammalian protein expression and characterization tools for next generation biologics next generation for tools characterization and expression protein Mammalian Doctoral Thesis in Biotechnology Mammalian protein expression and characterization tools for next generation biologics NIKLAS THALÉN ISBN TRITACBHFOU: KTH www.kth.se Stockholm, Sweden Mammalian protein expression and characterization tools for next generation biologics NIKLAS THALÉN Academic Dissertation which, with due permission of the KTH Royal Institute of Technology, is submitted for public defence for the Degree of Doctor of Philosophy on June 11th, 2021 at 10:00, F3, Lindstedsvägen 26, våningsplan 2, Sing-Sing, KTH campus, Stockholm. Doctoral Thesis in Biotechnology KTH Royal Institute of Technology Stockholm, Sweden 2021 © Niklas Thalén ISBN 978-91-7873-927-1 TRITA-CBH-FOU-2021:21 Printed by: Universitetsservice US-AB, Sweden 2021 Till Sofia Abstract Protein therapeutics are increasingly important for modern medicine. Novel recombinant proteins developed today can bind towards their target with high specificity and with low adverse effect. This has enabled the treatment of diseases that for a few years ago were deemed uncurable. Discovery of therapeutic proteins is driven through protein engineering, a field that is in constant expansion. And, through artificial construction of recombinant proteins, a large array of diseases can be defeated. The function and quality of these protein therapeutics rely on the correct folding, assembly and residue modification that occurs during their production within a living production cell host. Furthermore, producing them in large quantities are essential for accessibility of the best biopharmaceuticals available. Commonly, mammalian cells are the production host of choice when it comes to production of biopharmaceuticals. -

Structural and Numerical Changes of Chromosome X in Patients with Esophageal Atresia
European Journal of Human Genetics (2014) 22, 1077–1084 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg ARTICLE Structural and numerical changes of chromosome X in patients with esophageal atresia Erwin Brosens*,1,2,5, Elisabeth M de Jong1,2,5, Tahsin Stefan Barakat3, Bert H Eussen1, Barbara D’haene4, Elfride De Baere4, Hannah Verdin4, Pino J Poddighe1, Robert-Jan Galjaard1, Joost Gribnau3, Alice S Brooks1, Dick Tibboel2 and Annelies de Klein1 Esophageal atresia with or without tracheoesophageal fistula (EA/TEF) is a relatively common birth defect often associated with additional congenital anomalies such as vertebral, anal, cardiovascular, renal and limb defects, the so-called VACTERL association. Yet, little is known about the causal genetic factors. Rare case reports of gastrointestinal anomalies in children with triple X syndrome prompted us to survey the incidence of structural and numerical changes of chromosome X in patients with EA/TEF. All available (n ¼ 269) karyotypes of our large (321) EA/TEF patient cohort were evaluated for X-chromosome anomalies. If sufficient DNA material was available, we determined genome-wide copy number profiles with SNP array and identified subtelomeric aberrations on the difficult to profile PAR1 region using telomere-multiplex ligation-dependent probe amplification. In addition, we investigated X-chromosome inactivation (XCI) patterns and mode of inheritance of detected aberrations in selected patients. Three EA/TEF patients had an additional maternally inherited X chromosome. These three female patients had normal random XCI patterns. Two male EA/TEF patients had small inherited duplications of the XY-linked SHOX (Short stature HOmeoboX-containing) locus. -

Example Document Medical Genetics Center
รายงานผลการตรวจววเคราะหย เลขทธรออางอนง : 00000 ศผนยยพวนธธศาสตรยการแพทยย 6/6 หมนม 6 ถนนสสขาภนบาล 5 ซอย 32 แขวงออเงนน เขตสายไหม กรสงเทพมหานคร 10220 Accreditation No.4148/57 โทรศกพทย/โทรสาร 086-8861203, 02-7920726 e-mail : [email protected] www.genetics.co.th ชชชอ น.ส............................. HN. ............. โรงพยาบาล .................................... ววนททชเกกบตววอยยาง 19 พฤศจนกายน 2558 12:55 แพทยยผผผสวชงตรวจ นพ.................................... ววนททชรวบตววอยยาง 20 พฤศจนกายน 2558 12:43 ววธทการตรวจ G-banding ชนวดตววอยยาง นนนาครรนา ขผอบยงชทชในการตรวจ Elderly Pregnancy หนยวยสยงตรวจ ไมมมธ Center document Genetics ผลการตรวจExample: 47,XXX คคาอธวบาย : ตรวจพบโครโมโซม 47 โครโมโซม,เปปนเพศหญนง,พบโครโมโซม X เกนนมา 1 โครโมโซม เขอาไดอกกบ Triple X Syndrome Medical หมายเหตธ: Band level 450 band ขผอจคากวดในการทดสอบ: ไมมสามารถตรวจสอบความผนดปกตนขนาดเลลกชนนด submicroscopic rearrangement หรรอ low-level mosaicism ไดอ ผผผรวบรองผล ( .................................. ) ววนททชออกรายงาน : ....................................... นพ.วทรยธทธ ประพวนธยพจนย,พบ.,วว.กธมารเวชศาสตรย American Board of Medical Genetics (Clinical Cytogenetics) Medical Genetics Center : Fo-QP-19/01 : 16-03-2015 : 02 เรียน อ.นพ...................................... ที่นับถือ ตามที่ผลตรวจโครโมโซมจากน ้าคร่้าของ นส........................ (HN .............., Lab no. ..............) พบ โครโมโซม X เกินมา 1 โครโมโซมนั น ความผิดปกติดังกล่าว เข้าได้กับกลุ่มอาการ trisomy X หรือ triple X syndrome ซึ่งเป็นความผิดปกติของโครโมโซมเพศที่พบได้บ่อยในทารกเพศหญิง โดยความผิดปกติของ โครโมโซมแบบนี -

Identification of Differentially Expressed Genes in Human Bladder Cancer Through Genome-Wide Gene Expression Profiling
521-531 24/7/06 18:28 Page 521 ONCOLOGY REPORTS 16: 521-531, 2006 521 Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling KAZUMORI KAWAKAMI1,3, HIDEKI ENOKIDA1, TOKUSHI TACHIWADA1, TAKENARI GOTANDA1, KENGO TSUNEYOSHI1, HIROYUKI KUBO1, KENRYU NISHIYAMA1, MASAKI TAKIGUCHI2, MASAYUKI NAKAGAWA1 and NAOHIKO SEKI3 1Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; Departments of 2Biochemistry and Genetics, and 3Functional Genomics, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received February 15, 2006; Accepted April 27, 2006 Abstract. Large-scale gene expression profiling is an effective CKS2 gene not only as a potential biomarker for diagnosing, strategy for understanding the progression of bladder cancer but also for staging human BC. This is the first report (BC). The aim of this study was to identify genes that are demonstrating that CKS2 expression is strongly correlated expressed differently in the course of BC progression and to with the progression of human BC. establish new biomarkers for BC. Specimens from 21 patients with pathologically confirmed superficial (n=10) or Introduction invasive (n=11) BC and 4 normal bladder samples were studied; samples from 14 of the 21 BC samples were subjected Bladder cancer (BC) is among the 5 most common to microarray analysis. The validity of the microarray results malignancies worldwide, and the 2nd most common tumor of was verified by real-time RT-PCR. Of the 136 up-regulated the genitourinary tract and the 2nd most common cause of genes we detected, 21 were present in all 14 BCs examined death in patients with cancer of the urinary tract (1-7). -

Inside This Issue
Winter 2014 No. 77 Inside this issue Group News | Fundraising | Members’ Letters | One Family Living with Two Different Chromosome Disorders | Bristol Conference 2014 | Unique Leaflets | Christmas Card Order Form Sophie, Unique’s Chair of Trustees Dear Members, In the past month a few things have reminded me of why it is so important to make connections through Unique but also to draw support from other parents around us. I’ve just returned from Unique’s most recent family conference in Bristol where 150 of us parents and carers had a lovely time in workshops, meals and activities, chatting and watching our children milling around together like one big family since – although we had never met before – we have shared so many experiences in common. However in contrast I have also just met a new mum who has just moved to my area from far away with two toddlers, one with a rare joys of the internet, it is becoming easier to meet others with similar, chromosome disorder, who is starting from scratch with no even very rare, chromosome disorders around the world and to find professional, medical or social support. She reminds me of how yourself talking to them in the middle of the night about some lonely I felt when Max was newly diagnosed, when I knew no one interesting things our children share in common (obsession with with a disabled child let alone anyone with a rare chromosome catalogues, anyone?) And of course we also have an enormous disorder. Elsewhere our latest Unique Facebook group, Unique amount in common with so many parents of children with other Russia, is also just starting up – so far it includes just a small special needs or disabilities around us in our own communities who number of members sharing very different experiences to mine here will often be walking the same path as us. -
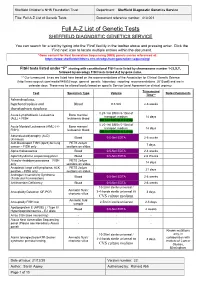
Full A-Z List of Genetic Tests Document Reference Number: 413.001
Sheffield Children’s NHS Foundation Trust Department: Sheffield Diagnostic Genetics Service Title: Full A-Z List of Genetic Tests Document reference number: 413.001 Full A-Z List of Genetic Tests SHEFFIELD DIAGNOSTIC GENETICS SERVICE You can search for a test by typing into the ‘Find’ facility in the toolbar above and pressing enter. Click the ‘Find next’ icon to locate multiple entries within the document. *Gene content for Next Generation Sequencing (NGS) panels can be referenced at: https://www.sheffieldchildrens.nhs.uk/sdgs/next-generation-sequencing/ FISH tests listed under “F” starting with constitutional FISH tests listed by chromosome number 1-22,X,Y, followed by oncology FISH tests listed A-Z by gene name. ** Our turnaround times are listed here based on the recommendations of the Association for Clinical Genetic Science (http://www.acgs.uk.com/media/949852/acgs_general_genetic_laboratory_reporting_recommendations_2015.pdf) and are in calendar days. These may be altered locally based on specific Service Level Agreement or clinical urgency. Turnaround Test Specimen Type Volume Notes/Comments Time** Achondroplasia, hypchondroplasia and Blood 0.5-5ml 2-6 weeks thanatophoric dysplasia 0.25-1ml BM in 5-10ml of Acute Lymphoblastic Leukaemia Bone marrow/ transport medium 14 days (ALL) + FISH leukaemic blood OR 1ml BM/VB in Li Hep 0.25-1ml BM in 5-10ml of Acute Myeloid Leukaemia (AML) (+/- Bone marrow/ transport medium 14 days FISH) leukaemic blood OR 1ml BM/VB in Li Hep Adrenoleukodystrophy (ALD) Blood 0.5-5ml EDTA 2-6 weeks (X-linked)